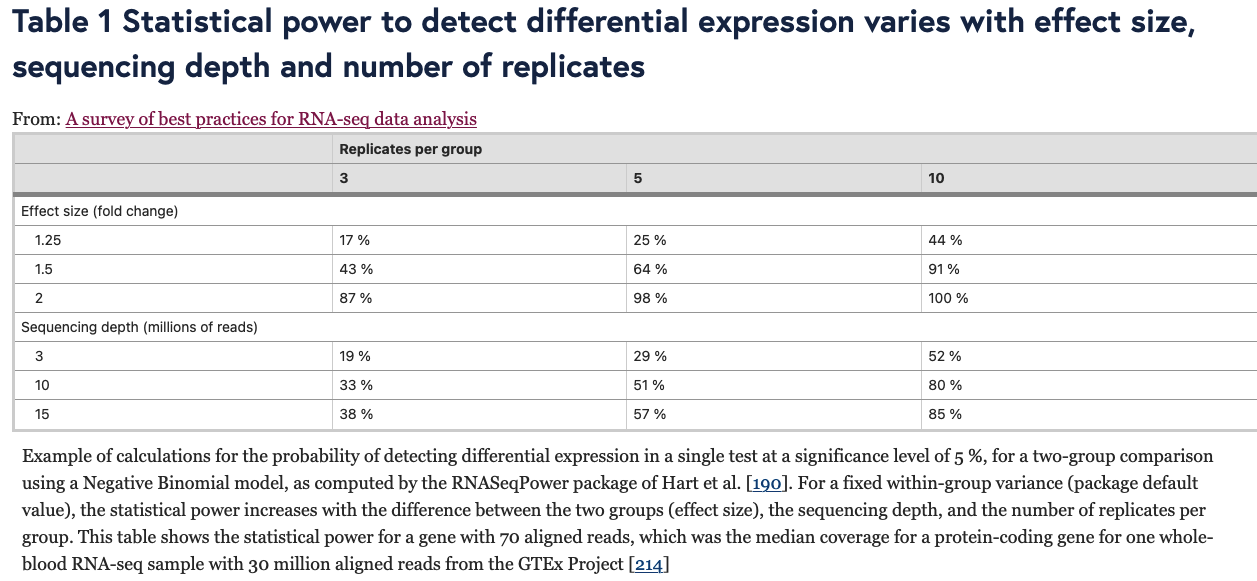
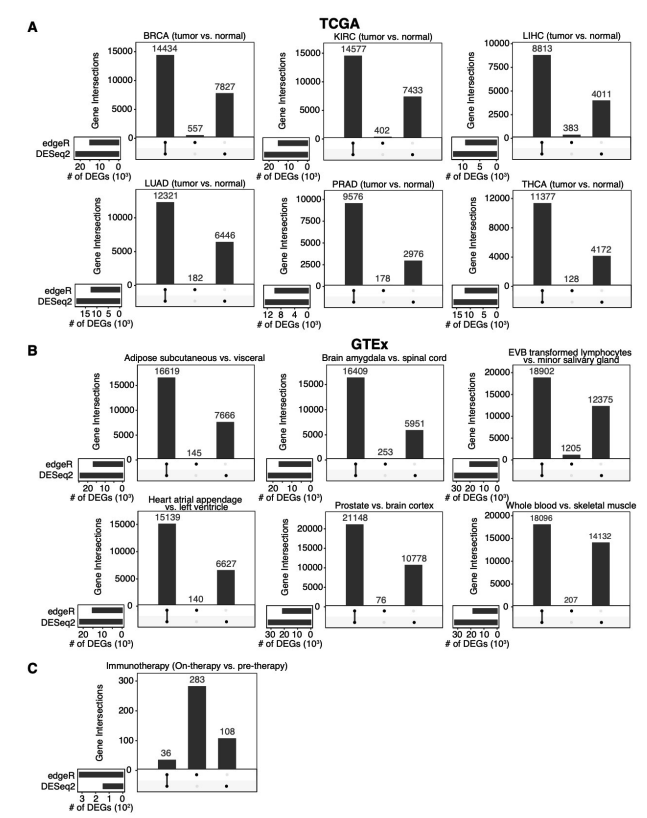
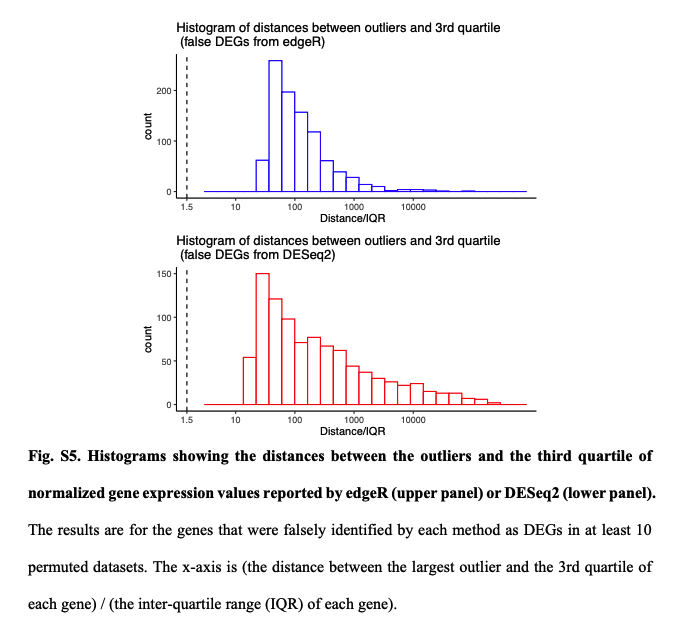
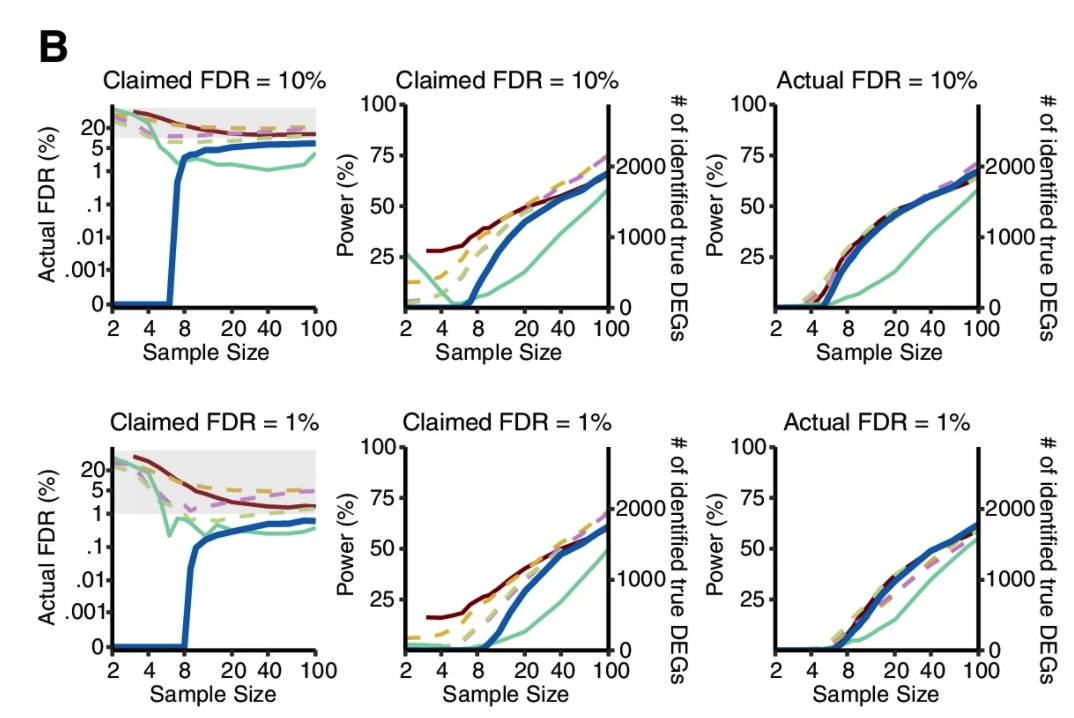
如无必要,勿增实体。样本够多,无需预设。 读博那几年,闲着没事就喜欢做各种软件的测试对比。有时候几个转录组样本非得用两三个差异分析方法都做一遍。严谨起来就给它们之间求一个交集,狡猾起来就谁的结果「更好」用谁(想必你也是这么做的)。 2021年,从植物研究转到肿瘤领域后,对于处理TCGA这类大样本队列,我其实不少次也是通过对tumor和normal组直接进行Wilcoxon秩和检验来找差异基因,原因无它,唯快不破。当然,更多时候是两种方法混用,好处是样本量多了跑上DESeq2可以离开座位溜达两圈活动活动。 2022年,前几天发表在 Genome Biology 的一篇论文,算是比较严谨地论证了在大样本量RNA-seq差异分析时,今后即便不考虑速度因素,也应该抛弃DEseq2和edgeR转而使用朴实无华的Wilcoxon秩和检验。 先贴上paper链接,你可以直接读原文或者继续看完这篇文章。 Li, Yumei, Xinzhou Ge, Fanglue Peng, Wei Li, and Jingyi Jessica Li. “Exaggerated False Positives by Popular Differential Expression Methods When Analyzing Human Population Samples.” Genome Biology 23, no. 1 (March 15, 2022): 79. https://doi.org/10.1186/s13059-022-02648-4. RNA-seq问世以来,最基本也是最关键的应用一直是在2组(有时候也有多组)情况下识别差异表达基因(DEG),比如肿瘤和正常组织样本,不同处理条件下的细胞系等等。 实际使用时,高通量的转录组数据我们通常只会测三个重复,这里的重复是指生物学重复而非技术重复。有的时候2个重复也能应付,甚至如果没有重复也有相关的软件可以处理。 为什么是3次重复呢?因为事不过三,以及材料有限和可以省钱。实际上,在不少早期专门研究应该有几次重复的论文中,大家的说法也不一,有说至少要6次才准的,也有说4次就够了,还有的说如果想把差异表达基因都找到至少需要12次个重复。 2016年,同样发表在Genome Biology的经典高引用RNA-seq分析综述则给出了如下表格和建议,简而言之,至少3次。 咱就是说,其实前期这些关于样本重复数量的分析,绝大多数都是在用DEseq和edgeR这些软件的基础上展开的。而今天却建议大样本量的分析不要再使用它们了,也蛮有趣。 从以上关于样本量的讨论可以看出,转录组差异分析最大的挑战一直是希望三生万物(三次重复得到普适结论),检测的样本量太少。 为了解决这个问题,通过对RNA-seq数据进行各种参数分布假设的统计方法和软件应运而生。其中最流行的两种方法是DEseq2和edgeR,他们的核心假设都是测序数据符合负二项分布,再根据自己的一套逻辑对原始读数进行标准化。如果样本没有重复,负二项分布可能有噪声,可以考虑基于柏松分布的DEGseq。 另一个重要的问题是,对于DEseq2和edgeR这样的软件,为了让各种后续处理成立,一个需要接受的假设是两组样本之间没有绝对意义上的差别,或者说绝大多数基因在两组之间的表达都是一样的。 于是就带来了第一个疑问,一旦上升到大样本量的人群队列RNA-seq研究,样本规模已经是大几十或者几百上千个,绝大多数基因的表达水平没有差别的这个假设是否还成立呢? 为了评估DESeq2和edgeR识别差异基因上的能力,这篇paper的作者测试了13个群体水平的RNA-seq数据集,总样本量从100到1376不等。分析后发现,DESeq2和edgeR在这些数据集上识别的DEG有很大的差异。 通过上图这个结果可以看出来在免疫治疗队列里,DESeq2和edgeR识别的DEG中只有8%一致。而在大多数大人群样本中,DESeq2都要相比edgeR找到的多不少。 事出反常必有妖,想看究竟问题出在哪里,就需要从FDR的角度进行深入的探索。测试FDR,在大样本的优势就体现出来了,它可以采取随机生成置换数据集的方法进行测试。 于是作者把队列中的两组样本进行混合,新的分组中每个都含有原来的两组样本。通过随机组合1000次置换,原始数据集就生成了1000次置换数据集。 结果很刺激。 首先,DESeq2和edgeR分别有84.88%和78.89%的概率从置换数据集识别出比原始数据集更多的DEG。 其次,DEseq2和edgeR分别从原始数据集中识别出的144个和319个DEG中,有22个(15.3%)和194个(60.8%)是从至少50%的置换数据集中识别出来的,而这些DEG其实都是假的。这个假阳性的结果实在是太高了。 最重要的是,它们还发现 fold change 越大的基因,越有可能从置换数据集中被鉴定出来。但在实际分析时,我们都会认为FC越大那基因的差异越大,它就越重要。而真实情况可能是这些基因本身并非差异表达基因。 这里多说一句,如果你好奇那些其实是假阳性的基因究竟是什么富集在什么功能呢?嗯……免疫相关。 为什么DESeq2和edgeR从这个免疫治疗数据集中发现了这么多假阳性?最直接的想法就是这些数据不再符合DESeq2和edgeR所假设的负二项分布。 为了验证这一假设,研究者选择了两组基因。其中一组是从≥20%的置换数据集中确定为DEG的基因;第二组是从≤0.1%的置换数据集中确定为DEG的基因。评估负二项式模型对每组基因的适应程度后可以发现确实第一组基因的模型拟合效果更差,这与这些基因是假阳性事实也一致。 进一步,为什么模型不适用了?作者检查了所有在至少10%的置换数据集中被误认为是DEG的基因,相对于假定的负二项模型,所有这些基因的实际测量值都存在异常值。 edgeR和DESeq2这样的参数检验中,假设是一个基因在两种条件下具有相同的平均表达量。所以分析结果会受到异常值存在的严重影响。相比之下,Wilcoxon秩和检验在有离群值存在时会表现的更好,因为它的假设是基因在一个条件下的表达量比它在另一个条件下的表达量高或低的可能相等。也就是Wilcoxon秩和检验更关注的是排序而非实际高低。 不止是DESeq2和edgeR,从上面的图可以看出作者还比较了其它几种代表性的分析方法,其中limma-voom和DESeq2、edgeR一样是参数估计;NOISeq和dearseq(最近发表的专门针对大样本量差异分析的工具)则和Wilcoxon秩和检验一样是非参数检验。 在比较谁能找到更少的假阳性差异基因时,DESeq2和edgeR明显败了,其它几个表现都还不错。如果比较它们找到更多真实的差异基因能力呢? 研究者从12个GTEx和TCGA数据集中各产生了50个半合成数据集(有已知的真实DEG和假DEG)。如下图,只有Wilcoxon秩和检验能在0.001%至5%的FDR阈值范围内可以持续控制真实的FDR。此外,比较了这六种方法在实际FDR条件下的功效,Wilcoxon秩和检验要优于其他五种方法。下图蓝色线是Wilcoxon秩和检验,黄色和紫色虚线是DEseq2和edgeR。 既然Wilcoxon秩和检验适合大样本的差异基因分析,关键问题就来到了多大样本量算大样本? 为了研究样本量如何影响六种分析方法的效果,研究者对每个数据集进行了下采样,分别得到了从2到100个的样本量的不同数据集。 从下图结果可以看出。在1%的FDR阈值下,当每个条件的样本量小于8时Wilcoxon秩和检验作为一个非参数检验方法你就别用了。同时,只要当每个条件的样本量超过8个以后,Wilcoxon秩和检验取得了与三种参数检验方法(DESeq2、edgeR和limma-voom)或其他非参数检验方法相当或更好的表现。 考虑到DEG占所有基因比例也可能会对结果产生影响,他们还生成了5中差异基因比例(1%、3%、5%、9%和20%)的数据集,评估结果也显示Wilcoxon秩和检验的FDR和功效也不差。 所以,从这个分析结果来看,当每个条件的样本量大于8个以后,别犹豫,直接上Wilcoxon秩和检验。 懂的都懂,DESeq2、edgeR和limma这三种参数检验方法在转录组研究中长期占主导地位。基本上所有大型研究的差异分析结果都是用它们做出来。但是这三种方法最初都是为了解决小样本量分析的问题。 在群体水平的研究中具有更大样本量(至少几十个),因此往往就不再需要进行参数假设。同时,由于样本量越多越可能存在几个异常值,它们违反假设后会导致P值变得不稳定,进而导致FDR无效。 最后一个问题,就是在实际分析中应该怎样合理地使用Wilcoxon秩和检验进行差异表达分析呢? 不同于DESeq2, edgeR和limma,作为一种非回归的方法,Wilcoxon秩和检验无法调整各种可能存在的混杂因素(比如测序深度的差别)。要想使用Wilcoxon秩和检验,研究者也推荐RNA-seq样本先进行标准化消除批次效应后再进行计算。 至于你是使用DESeq2的Relative Log Expression,还是使用edgeR的Trimmed Mean of M-values,或者是使用TPM,个人粗浅的认为问题不大(狡猾的你肯定会愿意都试试)。 最后是一段使用edgeR TMM + wilcox.test 分析的代码示例,来自于原文作者。 看到这里,个人感觉也没有必要对自己以前做过的事情产生太大自我怀疑,只是以后在做超过每组8个样本的差异分析时,别忘了也捎带试试Wilcoxon秩和检验。如果有人问你为啥有专门的DESseq2和edgeR不用,就把这篇paper丢过去。 当然,还有这个图。 reference 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。
RNA-seq实验应该有几个重复


DEseq2和edgeR不适用大样本
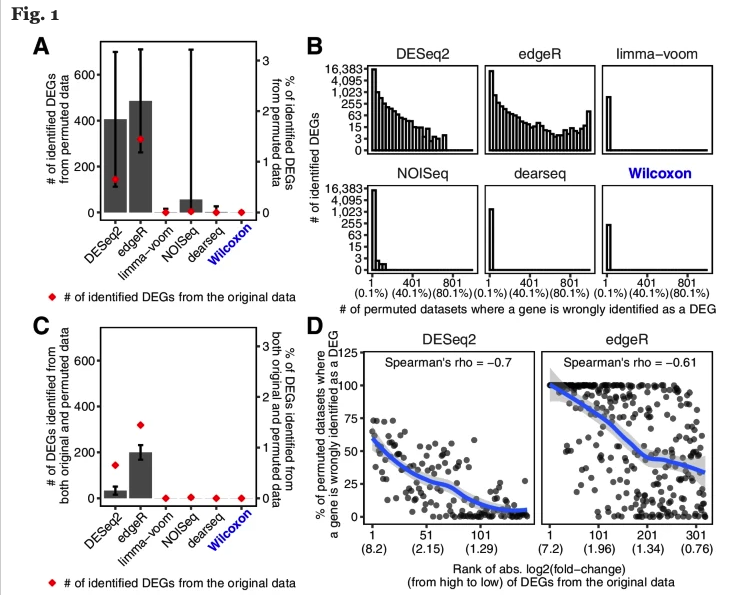
DEseq2和edgeR为什么不灵了
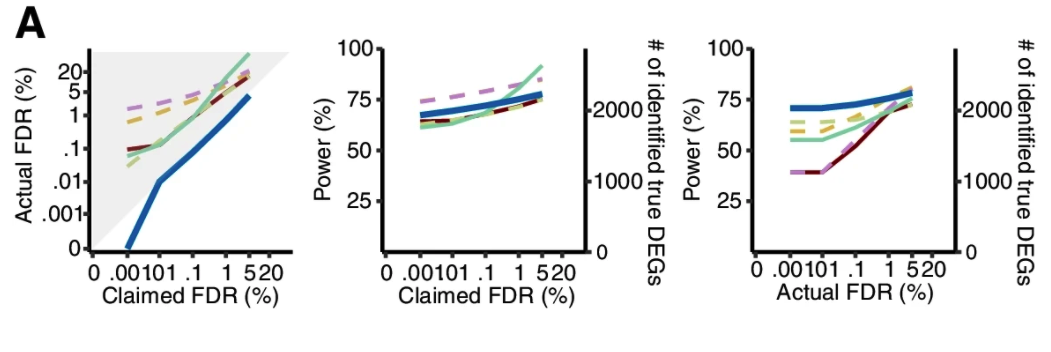
什么时候使用Wilcoxon秩和检验
怎么使用Wilcoxon秩和检验
# read data
readCount<-read.table(file="examples/examples.countMatrix.tsv", header = T, row.names = 1, stringsAsFactors = F,check.names = F)
conditions<-read.table(file="examples/examples.conditions.tsv", header = F)
conditions<-factor(t(conditions))
# edger TMM normalize
y <- DGEList(counts=readCount,group=conditions)
##Remove rows conssitently have zero or very low counts
keep <- filterByExpr(y)
y <- y[keep,keep.lib.sizes=FALSE]
##Perform TMM normalization and transfer to CPM (Counts Per Million)
y <- calcNormFactors(y,method="TMM")
count_norm=cpm(y)
count_norm<-as.data.frame(count_norm)
# Run the Wilcoxon rank-sum test for each gene
pvalues <- sapply(1:nrow(count_norm),function(i){
data<-cbind.data.frame(gene=as.numeric(t(count_norm[i,])),conditions)
p=wilcox.test(gene~conditions, data)$p.value
return(p)
})
fdr=p.adjust(pvalues,method = "fdr")
# Calculate fold-change for each gene
conditionsLevel<-levels(conditions)
dataCon1=count_norm[,c(which(conditions==conditionsLevel[1]))]
dataCon2=count_norm[,c(which(conditions==conditionsLevel[2]))]
foldChanges=log2(rowMeans(dataCon2)/rowMeans(dataCon1))
# Output results based on FDR threshold
outRst<-data.frame(log2foldChange=foldChanges, pValues=pvalues, FDR=fdr)
rownames(outRst)=rownames(count_norm)
outRst=na.omit(outRst)
fdrThres=0.05
write.table(outRst[outRst$FDR<fdrThres,], file="examples/examples.WilcoxonTest.rst.tsv",sep="\t", quote=F,row.names = T,col.names = T)
写在最后

· 分享链接 https://kaopubear.top/blog/2022-03-20-donot-use-deseq2-edger-in-human-population-samples/