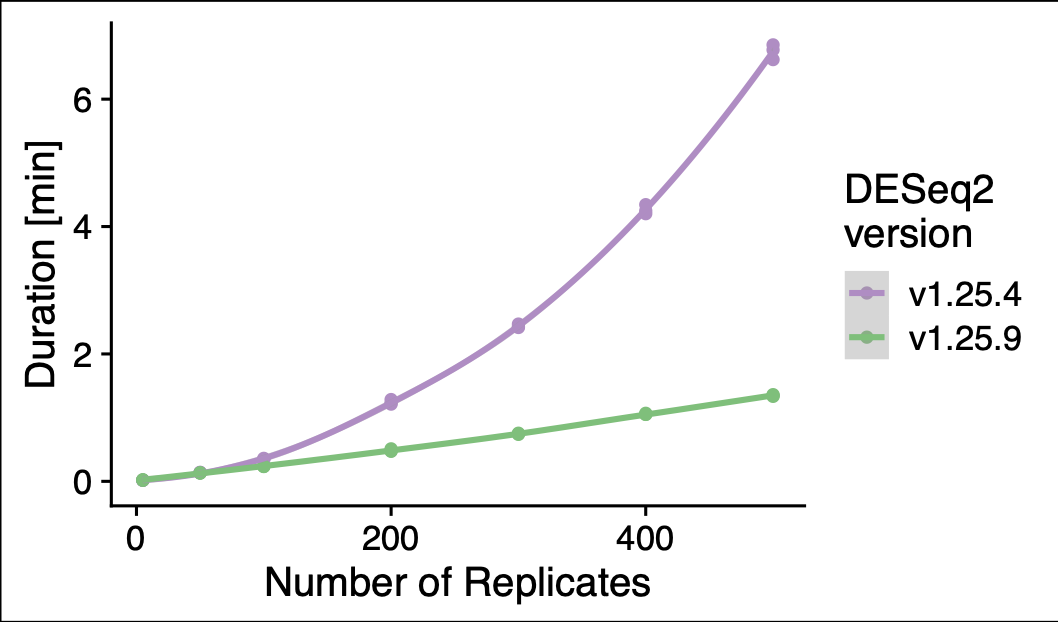
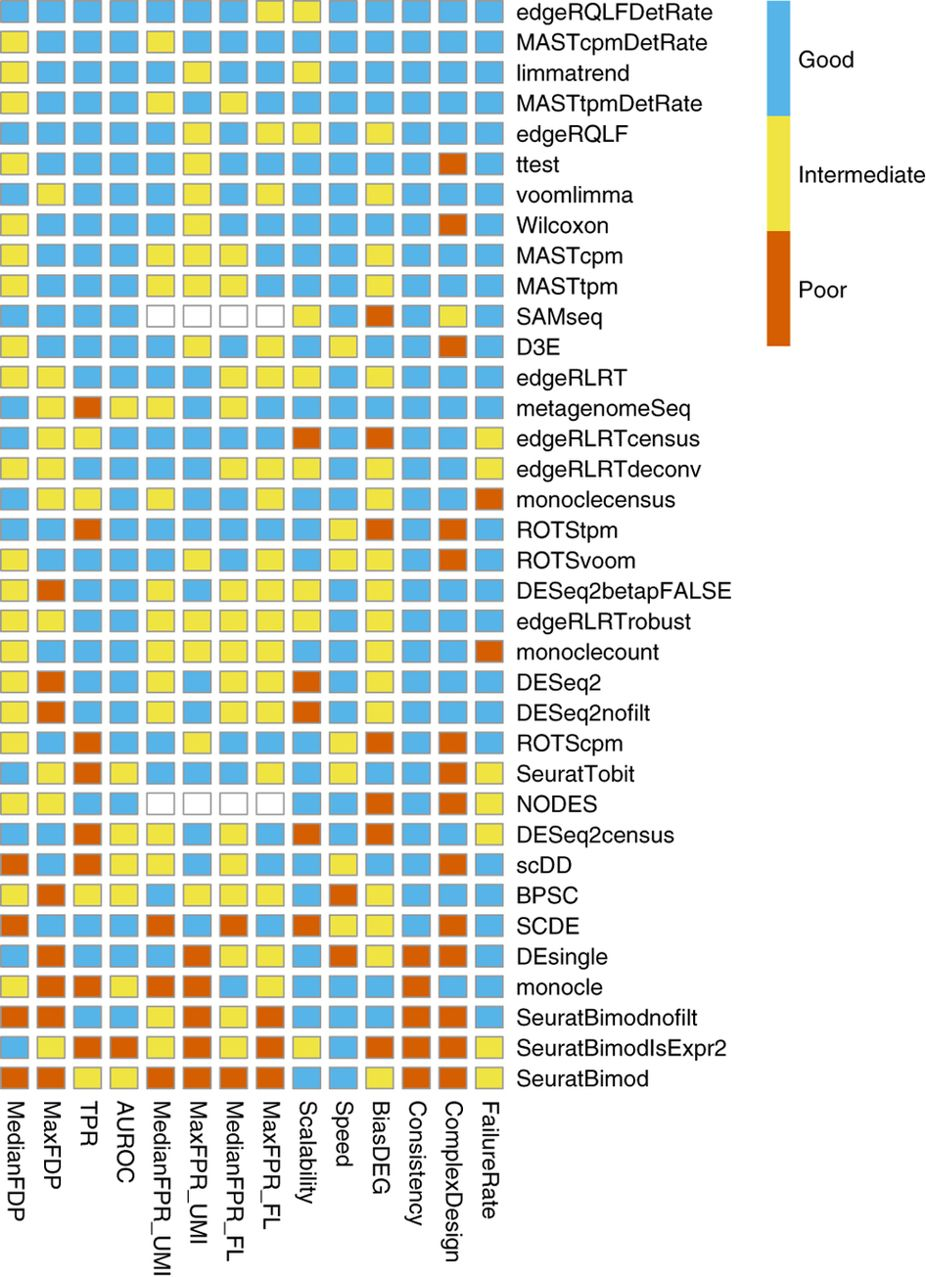
DESeq 最新版本(v1.25.9)针对大样本分析的速度和之前相比有了质的飞越,果子老师的 1215 个 TCGA 样本差异分析时间从 1200 分钟缩短到单线程 20 分钟,使用多线程的情况下最快 6 分钟搞定。 前一段时间洲更用统计学中的非参数检验方法解决了果子老师关于 1200 个 TCGA 样本使用 DESeq2 分析耗时 20 小时的问题,然后在那篇文章的最后果子老师如此总结道: 这样看来,洲更说的是对的。不过,dds 这里还有一个比较好的点就是可以做 logFC 矫正,就像这个帖子里面提到的,有一些 count 比较小,但是变化值很大的基因,会对 GSEA 分析产生影响。 从这段话里可以看出他还是心心念念 DEseq2。念念不忘必有回响,DEseq2 开发者听到了果子老师心里的呐喊,最近一次 GitHub 上 DEseq2 的更新就着手解决了这个问题。这段代码的贡献者称他们解决了 DEseq2 处理大量样本异常耗时问题,时间随样本量的变化趋势目前已经做到了线性关系。 对比图如下: 究竟效果如何,接下来就用果子老师 1215 个样本数据去测试。 第一次测试环境我选择了自己的台式机,16G 内存。不知道是出于对开发者的信任还是对电脑的信任或者是出于对果子老师代码的信任。我选择了直接复制了果子老师的代码开启并行模式进行分析。 在果子老师的当年的描述中,如此说道: 最困难的一步来了。燃烧你的小电脑。这里还使用了并行化处理来解决速度的问题。但是,耗时依然很长,4 个小时以上,有可能是 10 个小时 (后来,据我私下和果子老师交流,他说实际运行时间远远高于 4 小时,为了显示出自己机器性能的优越稍微在写作手法上做了一些加工,人为缩短了这个时间) 这里我并不知道果子老师小电脑的性能如何以及用了几个线程,姑且按照 4 个小时(240 分钟)来计算。首先要从 GitHub 安装最新版本的包,安装好之后查看包的版本是 1.25.9,没有问题。 以下是测试步骤,原始的 Rdata 来自果子老师,用构建好的 dds 直接做 DESeq 虽然只是一个函数其实包含了若干个步骤,其中耗时很严重的是 然后我就看了眼电脑的任务管理器,如下图所示。 接下来,就出问题了。因为 6 个线程工作再加上电脑只有 16G 内存, 事已至此,勿忘初心,迁移到服务器上去试试。在服务器安装最新版本的 DEseq2 包,继续运行同样内容,并行开启后,这次自动给我分配了 36 个线程,真棒! 接下来一切正常,运行时间如下所示: 全程跑完一共用时 6 分钟多一点,和之前果子老师的多线程 4 个多小时的时间比,速度提升了应该有 40 多倍的样子。这里再次强调一下,据我私下和果子老师交流实际运行时间远远要高于 4 小时哦。如此看来,文章开头的运行时间对比图还比较靠谱,线性时间的复杂度大大缩短了处理大量样本的时间。 随后,测试如果只开启一个线程的情况(现在很少有单线程的机器,但是不少人都不知道 DEseq 可以多线程运行),如果不用最新版的话,据一些学员反映这个时间是在 20 到 30 个小时之内,如果用了最新版呢?单线程整个时间缩短到 22 分钟左右。(从一个侧面来说,这里的多线程加速效果也并不是非常明显。) 从下文会提到的开发者更改内容来看,这次更新提速并没有更改涉及到 DEseq2 模型和参数的内容,所以新老版本的分析结果是完全一致的,包括差异基因的个数和 p 值。 回到文章开头,这次的升级当然不是听到果子老师远在他乡用微信公众号发出的呼唤,而是开发者为了让 DEseq2 可以更好的处理单细胞数据而准备的。在传统的差异分析问题上,我们面对的是高深度和少量几个样本,但是在单细胞上,面对的是上千个细胞和相对很浅的测序深度。 代码贡献者在自己的博客中提到,在几年前大家都还在争论转录组测序数据中大量 count 为 0 的数据应该如何处理,但是最近已经有阴性对照的实验数据表明这些 0 并不会有什么问题。另外,已经有文章测试表明,传统差异分析的很多工具在单细胞数据中依旧可以使用,甚至要比一些专门为单细胞开发的工具表现要好。如下图所示: 但是 DESeq2 开发者的实验室发现,DESeq2 和其他一票软件相比,在处理大量细胞时速度上完败,细胞越多败的越彻底。作为差异分析工具的大哥,当然也是要面子的。于是决定找出问题进行优化。 对于代码改动,可以从 GitHub 的 PR 中略知一二。 以下是 R 脚本的一个优化示例: 在 新版本中对应的代码如下图所示: 单独看几行代码可能有些难以理解,在这里尽力解释一下。 上面代码提到的 其中的参数 果子老师的数据中 实际运行 如下图所示,第二列 原始的 ModelMatrix 中第二列的样本情况 而 这个参数,可能大多数人都不会被注意到。他的含义是 the minimum number of replicates required in order to use replaceOutliers on a sample. 那么 书接上文, 扯远了,再说回来, 到这里很可能你已经看不下去了,但是其实不难理解。 原始函数使用 如果变成新的函数会有什么效果呢? 首先给 但是只要进行一次运算, 以果子老师的数据为例, 他只有两组数据,1215 个样本。只需要进行一次运算,完成相当于原始函数的 1102 次计算(癌有 1102 个),然后循环到下一个是 NA 的样本时(也就是第一个正常样本时),再进行一次运算,就完成了原始函数的 113 次计算。 也就是当面对 2000 个样本的 2 组数据时,原始函数需要运行 2000 次,而新的函数只需要运行 2 次。 在 1215 个样本的 2 组数据中,前者需要 3.17s,而后者只需要 0.03s。 当然,直接导致时间复杂度改变的升级是在核心的 C 脚本中,我也只能勉强读懂一些。 在原始的 但在新的代码中,避免了对于这个矩阵的计算。取代构造矩阵而是首先构造了一个 n 维向量,接下来不需要对全部矩阵进行计算就也可以得到对角线矩阵。这一步上的时间复杂度,就从之前的 Bias, robustness and scalability in single-cell differential expression analysis 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。太长不看版本
前奏
台式机多线程测试
DESeq。devtools::install_github("mikelove/DESeq2@ae7c6bd")
library(DESeq2)
library(BiocParallel)
dds <- DESeq(dds,parallel = T)
gene-wise dispersion estimates,这里也是多线程开启的地方。R 自动在我的台式机上开启了 6 个线程。起初我非常开心,新版本gene-wise dispersion estimates 这一步很快完成,目测只有不到 5 分钟。看情况不出问题的话,全部分析应该会在 10 分钟左右完成。
fitting model and testing到一半时我的心态伴随着内存一起都崩了。到崩溃为止,一共运行了大约 11 分钟。> dds <- DESeq(dds,parallel = T)
estimating size factors
estimating dispersions
gene-wise dispersion estimates: 6 workers
mean-dispersion relationship
final dispersion estimates, fitting model and testing: 6 workers
error: arma::memory::acquire(): out of memory
Error: BiocParallel errors
element index: 4
first error: std::bad_alloc
In addition: There were 13 warnings (use warnings() to see them)
> proc.time() - t1
user system elapsed
42.21 253.32 702.48
服务器多线程测试
> dds <- DESeq(dds,parallel = T)
estimating size factors
estimating dispersions
gene-wise dispersion estimates: 38 workers
mean-dispersion relationship
final dispersion estimates, fitting model and testing: 38 workers
-- replacing outliers and refitting for 9759 genes
-- DESeq argument 'minReplicatesForReplace' = 7
-- original counts are preserved in counts(dds)
estimating dispersions
fitting model and testing
> proc.time() - t1
user system elapsed
1720.880 207.212 379.153
单线程测试
> dds <- DESeq(dds,parallel = F)
estimating size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
-- replacing outliers and refitting for 9759 genes
-- DESeq argument 'minReplicatesForReplace' = 7
-- original counts are preserved in counts(dds)
estimating dispersions
fitting model and testing
> proc.time() - t1
user system elapsed
1369.724 18.084 1387.730
分析结果完全一致
contrast <- c("sample","cancer","normal")
dd1 <- results(dds, contrast=contrast, alpha = 0.05)
library(dplyr)
library(tibble)
library(tidyr)
### 导出差异分析的结果
res <- dd1 %>%
data.frame() %>%
rownames_to_column("gene_id")
> table(res$padj < 0.01)
FALSE TRUE
20502 25189
究竟改了什么
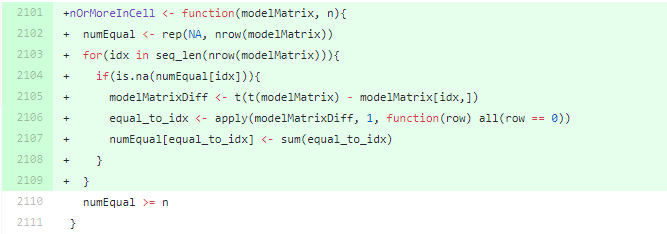
core.R中,旧版本的代码如下图所示
modelMatrix 参数来自于getModelMatrix()这个函数,内容如下:getModelMatrix <- function(object) {
if (is(design(object), "matrix")) {
design(object)
} else if (is(design(object), "formula")) {
stats::model.matrix.default(design(object), data=as.data.frame(colData(object)))
}
}
object就是我们实际数据中的 dds,这里首先判断 design(object) 的数据类型,以果子老师的数据为例。> is(design(dds))
[1] "formula" "oldClass"
design(object) 是 formula,那么就会运行stats::model.matrix.default(design(object), data=as.data.frame(colData(object)))来得到 ModelMatrixgetModelMatrix()我们得到的 modelMatrix其实是一个 1215*2 的矩阵。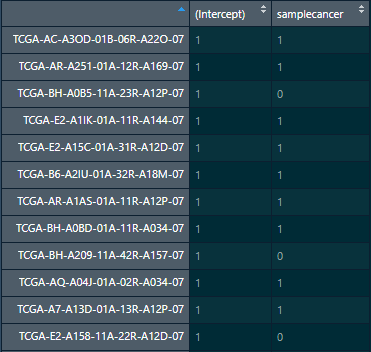
samplecancer就是分组信息,其中 0 代表正常,1 代表癌。metadata 中的样本数量如下> table(metadata$sample)
normal cancer
113 1102
> table(modelMatrix[,2])
0 1
113 1102
nOrMoreInCell函数的作用是返回一个每个样本一一对应的逻辑向量,来表明是否需要被筛选掉。这里又需要提到一个参数minReplicatesForReplace。replaceOutliers()又是做什么的呢?我们说 DEseq 在进行标准化的时候会丢掉一些 gene counts 数非常异常的值,转而使用所有样本中的 trimmed mean 来进行替代。replaceOutliers()就是做这件事的。minReplicatesForReplace 就指定了在对一个样本使用 replaceOutliers 的时候需要最小的重复数是多少。假如这个值是 5,如果你的一个样本只有 3 个重复,那么就不会执行上面的操作。这个参数默认是 7,假设你不想做这一步(例如针对单细胞数据),你就需要把这个参数设置为 Inf。在实际运行中,如何判断哪个样品该进行哪些样品不该进行这些操作呢?就是进行如下一个比较,下文代码的第一行也展示了nOrMoreInCell()的作用。 # if there are sufficient replicates, then pass through to refitting function
sufficientReps <- any(nOrMoreInCell(attr(object,"modelMatrix"),minReplicatesForReplace))
if (sufficientReps) {
object <- refitWithoutOutliers(object, test=test, betaPrior=betaPrior,
full=full, reduced=reduced, quiet=quiet,
minReplicatesForReplace=minReplicatesForReplace,
modelMatrix=modelMatrix,
modelMatrixType=modelMatrixType)
}
nOrMoreInCell() 是如何在新版本中提速的。nOrMoreInCell_old <- function(modelMatrix, n) {
numEqual <- sapply(seq_len(nrow(modelMatrix)), function(i) {
modelMatrixDiff <- t(t(modelMatrix) - modelMatrix[i,])
sum(apply(modelMatrixDiff, 1, function(row) all(row == 0)))
})
numEqual >= n
}
sapply在每个样品上都要迭代一次,因此如果的设计矩阵很简单就会出现大量的重复工作。对于大量样本这个函数占用 DESeq()相当一大部分运行时间。对于 1200 个样本的数据来说,这个过程要重复 1200 次才能得到结果。nOrMoreInCell_new <- function(modelMatrix, n){
numEqual <- rep(NA, nrow(modelMatrix))
for(idx in seq_len(nrow(modelMatrix))){
if(is.na(numEqual[idx])){
modelMatrixDiff <- t(t(modelMatrix) - modelMatrix[idx,])
equal_to_idx <- apply(modelMatrixDiff, 1, function(row) all(row == 0))
numEqual[equal_to_idx] <- sum(equal_to_idx)
}
}
numEqual >= n
}
numEqual 全部复制为NA;当进行第一次循环idx=1时,因为numEqual[idx]是NA,所以一定会执行一次运算。equal_to_idx 就会返回一个新的逻辑值,所有equal_to_idx为真的位置对应的numEqual的位置都会被赋值这个样本重复的数量,也就是sum(equal_to_idx)。然后第二次循环开始,对于和第一次循环同样组的样本来说,只需要进行if(is.na(numEqual[idx])) 的判断,因为必定会返回False 就不需要再进行后续运算了。> t1 <- proc.time()
> samplesForCooks <- nOrMoreInCell_new(modelMatrix,3)
> proc.time() - t1
user system elapsed
0.00 0.02 0.03
>
> t1 <- proc.time()
> samplesForCooks <- nOrMoreInCell_old(modelMatrix,3)
> proc.time() - t1
user system elapsed
3.06 0.08 3.17
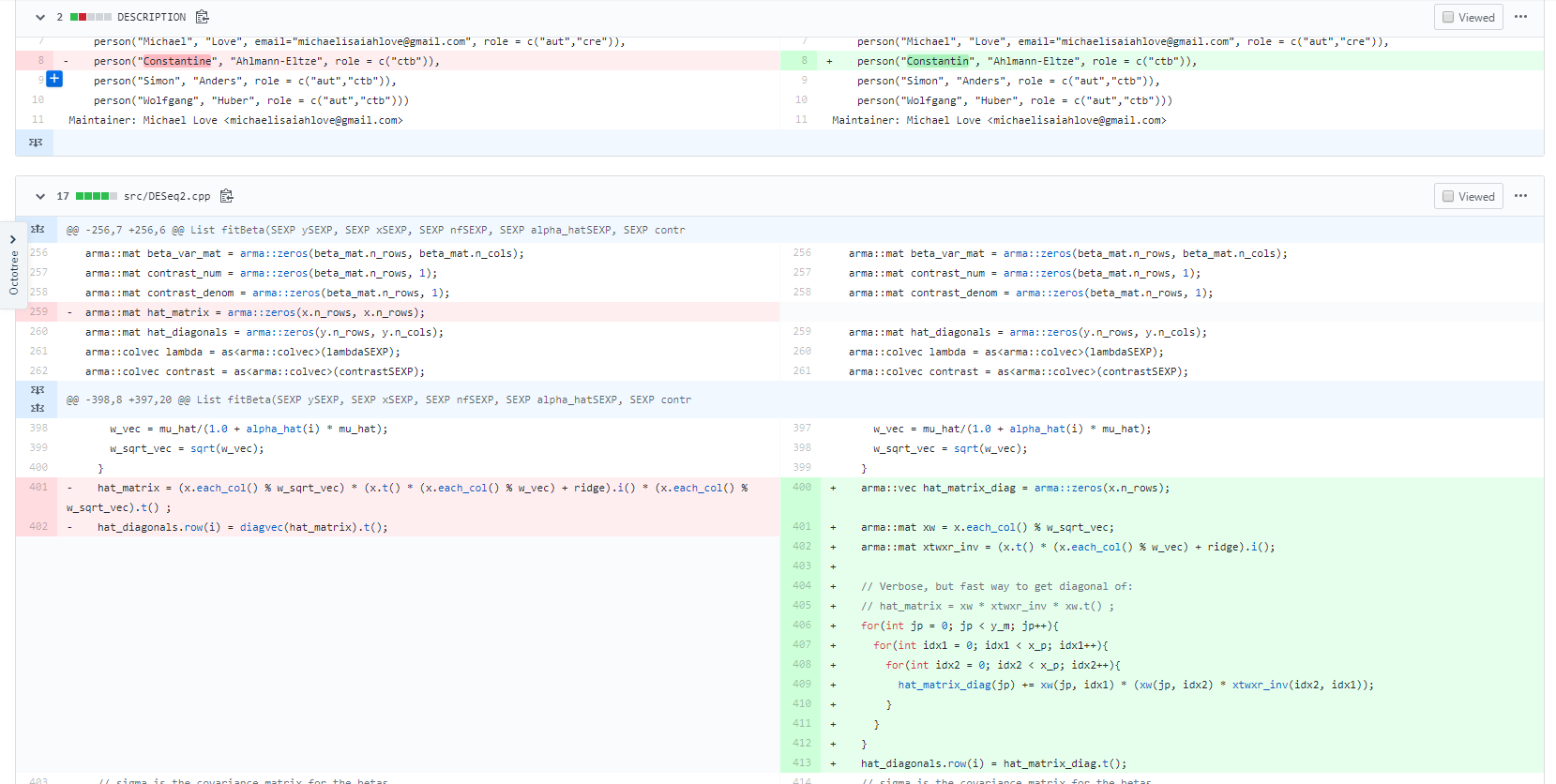
DESeq2.cpp中有一步需求是提取对角线矩阵。原代码首先构造了一个 n 行 n 列的矩阵,然后再对整个矩阵进行运算,最后再用diagvec()来提取对角线矩阵,这个思路没什么问题,但是随着样本数量的增加,对于矩阵的运行时间是成样本数的平方增加的,遇到大量样本直接歇菜。O(ncol(Y)^2) 变成了 O(ncol(Y))的线性复杂度。这也是我们看到文章开始运行时间变化的主要原因。进一步了解
· 分享链接 https://kaopubear.top/blog/2019-09-17-fasterdeseq2/