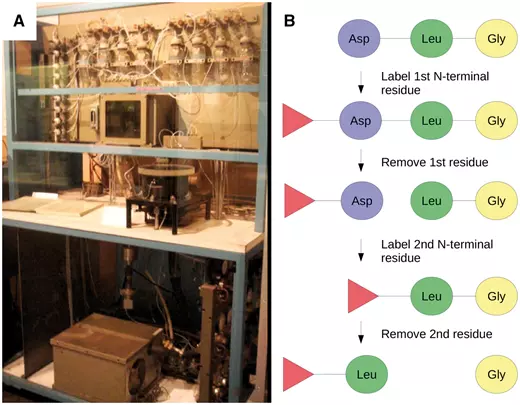
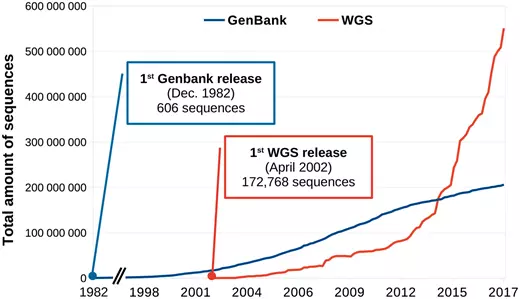
前几天看到 Briefings in bioinformatics 发了一篇 文章 介绍生物信息学发展历史。个人有两个感慨,一是这样的文章也可以发表(不禁想给自己的博客投稿),二是感慨生物信息学发展之快。对整篇文章进行了粗糙的翻译整理,供大家了解。 20 世纪 50 年代早期,DNA 的历史地位还没有被建立,那个时候人们普遍认为蛋白质才是遗传信息的载体。直到 1952 年的噬菌体感染实验,人们才第一次证明 DNA 是真正的遗传物质。因为这个历史原因,生物信息学在 DNA 中的应用要落后于蛋白质研究将近 20 年时间。 50 年代后期,人们得到了胰岛素的蛋白质序列,这一成就激励人们去开发获得蛋白质序列更有效的方法。Edman 降解法就是其中之一,肽链的第一个 N-末端氨基酸用异硫氰酸苯酯(PITC)标记,然后通过降低 pH 来进行切割。通过重复该过程,一次一个 N-末端氨基酸进而可以确定肽序列。 Margaret Dayhoff(1925-1983)是一位美国物理化学家,他开创了计算方法在生物化学领域的应用。Dayhoff 对这一领域的贡献非常重要,NCBI 前主任 David J. Lipman 称她为”生物信息学的母亲和父亲” 。1960 年,她成为国家生物医学资源基金会的副主任。与 Robert S. Ledley 一起开发出了第一个生物信息学软件:COMPORTEIN , 用于使用 Edman 测序数据确定蛋白质的一级结构,使用 fortran 语言开发并运行在打孔卡上,完全运行在这个软件运行在 IBM 7090 大型机上,如下图 A 所示。 在 COMPROTEIN 软件中,输入和输出氨基酸序列以三个字母的缩写表示(例如赖氨酸的 Lys,丝氨酸的 Ser)。为了简化蛋白质序列数据的处理,Dayhoff 后来开发了目前仍在使用的单字母氨基酸代码。这个单字母代码最初用于 Dayhoff 和 Eck 的 1965 年蛋白质序列和结构图谱,而它是有史以来第一个生物序列数据库。第一版包含 65 种蛋白质序列,其中大部分是少数蛋白质的种间变异。 1970 年,Needleman 和 Wunsch 开发了第一个成对蛋白质序列比对的动态编程算法,80 年代早期,做为 Needleman-Wunsch 算法的推广,第一个多序列比对(MSA)算法首次公布,但是这个算法并没有太大的价值,因为其时间复杂度是,L 代表序列长度,N 代表序列数量。找到最佳比对的时间与序列长度的序列数量次幂成正比。 多序列比对第一个真正成熟的算法由 Da-Fei Feng 和 Russell F. Doolitle 于 1987 年开发,流行的 MSA 软件 CLUSTAL 开发于 1988 年,作为 Feng-Doolittle 算法的简化至今仍在使用和维护。 ##蛋白质到 DNA: 1970-1980 1968 年,64 个密码子都被解析出来,DNA 成为可读信息后要求我们能都快速获得 DNA 序列。 第一种被广泛采用的 DNA 测序方法是 1976 年发表的 Maxam-Gilbert 测序方法。由于该方法使用了放射性和危险化学品, 同时其固有的复杂性在很大程度上阻碍了其用于支持 Frederick Sanger 实验室开发的方法。Sanger 团队在 1977 年开发出名为“plus and minus” 的 DNA 测序方法,这是第一个要依赖 DNA 聚合酶进行而成的方法,对该方法进行改进后诞生了目前常见的 Sanger 双脱氧链终止法 ,即使在成立 40 年后的今天它仍在广泛使用中。 第一个专门用于分析 Sanger 测序读数的软件由 Roger Staden 于 1979 年发表。该程序集可分别用于 Staden Package 是第一个包含附加字符(Staden 称为“不确定代码”)的序列分析软件,以用于记录序列读取中的非确定性碱基。这种扩展的 DNA 字母表是现代 IUBMB(国际生物化学和分子生物学联合会)命名法的前身之一,用于核酸序列中未完全指定的碱基。 Felsenstein 是第一个利用最大似然(ML)方法从 DNA 序列推断系统发育树的软件。在 Felsenstein 之后已经开发出了若干种使用 ML 的生物信息学工具以及用于评估节点稳健性的新统计方法。从某种程度上,在 20 世纪 90 年代人们因此受到启发,将贝叶斯统计应用在分子系统发育中并使用至今。 在 20 世纪 70 年代后半期,人们又必须克服一些技术限制以扩大计算机在 DNA 分析中的应用(更不用说 DNA 分析本身)。接下来的十年是解决这些问题的关键。 与蛋白质和 RNA 不同,基因不能进行生物化学分级然后单独测序,因为它们都是连续地在每个细胞的少量 DNA 分子上连续存在。基因通常以每个细胞一个或几个拷贝存在,基因的数量级比它们编码的产品的数量级少很多,可以说 DNA 是可以测序的最不丰富的大分子细胞成分。 1972 年当 Jackson,Symons 和 Berg 使用限制性核酸内切酶和 DNA 连接酶切割并将环状 SV40 病毒 DNA 插入 λDNA 并用该载体转化大肠杆菌细胞时,该问题得到了部分解决。插入的 DNA 分子在宿主生物中复制时也随着大肠杆菌培养物的生长而扩增,产生出数百万拷贝的单个 DNA 插入物。该实验开创了独立于来源生物的基因的分离和扩增技术。然而,因为 Berg 非常担心这其中存在的伦理问题,因为他曾要求暂停使用 DNA 重组技术,并在 1975 年组织建立了一系列用于现代遗传学实践的指南。 操纵 DNA 的第二个里程碑是聚合酶链式反应(PCR)的出现,它允许在没有克隆程序的情况下扩增 DNA。尽管使用 DNA 聚合酶进行"修复合成"的描述是在 1971 年由 Kjell Kleppe 等人首次提出的。但 PCR 的发明却要归功于 Kary Mullis,因为他对该方法进行了大量优化,特别是使用热稳定 Taq 聚合酶,以及开发热循环仪。 基因克隆和 PCR 现在常用于 DNA 文库制备,这对于获得序列数据至关重要。20 世纪 70 年代后期 DNA 测序的出现,以及增强的 DNA 操作技术,已经产生了越来越多的可用序列数据。与此同时,20 世纪 80 年代,人们开始越来越多地使用计算机和相关生物信息学软件。 在 20 世纪 70 年代之前, 所谓的“小型计算机”仍然相当于一台小型家用冰箱的尺寸和重量(如下图所示),而且还不包括终端和存储单元。这样的体积限制使得个人或小型工作组的计算机购置变得异常繁琐。而且使用起来也异常的不友好。 第一波即用微型计算机于 1977 年进入消费市场。这一波浪潮出现的首批计算机包括三种型号:Commodore PET,Apple II 和 Tandy TRS-80,在比试,它们代表着体积小,价格低廉和用户友好。这三种计算机都具有一个内置的 BASIC 解释器,这 对于非程序员而言是一种简单的语言。 生物学计算机软件亦开始迅速发展。1984 年,威斯康星大学遗传学计算机课题组 (Genetics Computer Group) 发表了与他们同名的“GCG”软件合集。GCG 软件包是包括 33 个命令行工具的集合,可以用于操作 DNA,RNA 或蛋白质序列。要记住,这是为序列分析开发的第一个软件集合。 1985 年,Richard Stallman 发表了 GNU 宣言,概述了他创建名为 GNU(基于 Unix)免费操作系统的动机。这一运动后来成长为自由软件基金会 ( Free Software Foundation ),该基金会倡导”用户可以自由运行,复制,分发,研究,改变和改进软件”的理念。 Stallman 倡导的自由软件哲学是生物信息学的若干倡议的核心,其它的倡议还包括如欧洲分子生物学开放软件套件,其发展始于 1996 年,作为自由和开放源码用来替代 GCG。 最重要的是,在此期间欧洲分子生物学实验室(EMBL),GenBank 和日本 DNA 数据库(DDBJ)开始联合起来(1986 年 EMBL 和 GenBank 加入,1987 年 DDBJ 加入)。为了规范化数据格式定义了报告核苷酸序列的最简信息,并促进数据库之间数据共享。 20 世纪 80 年代也是生物信息学在现代科学中存在感足以获得专门期刊的时刻。鉴于计算机可用性大幅提高以及在生物领域进行计算机辅助分析的巨大潜力,专门针对生物信息学的期刊* Computer Applications in the Biosciences(CABIOS)* 于 1985 年成立,现在这个期刊已经更名为为* Bioinformatics*。 自由软件运动和专用科学期刊的出现拓宽了生物学中计算机的使用范围。然而,对于诸如全基因组和基因目录的大型数据集,使用小型大型计算机而不是微型计算机。这些系统通常在类 Unix 操作系统上运行,并且使用不同于微型计算机上常用的编程语言(例如 C 和 FORTRAN)(例如 BASIC 和 Pascal)。因此,为微型计算机制造的流行序列分析软件并不总是与大型计算机兼容,反之亦然。 随着 20 世纪 80 年代早期 x86 和 RISC 微处理器的出现,出现了一类新的个人计算机。桌面工作站专为技术和科学应用而设计,具有与微型计算机相当的尺寸,但具有更高的硬件性能,以及更类似于大型计算机的软件架构。事实上,桌面工作站通常运行在 Unix 操作系统和衍生产品上,如 HP-UX 和 BSD(图 3)。 20 世纪 80 年代中期出现了几种脚本语言,这些语言在今天的生物信息学家中仍然很受欢迎。这些语言抽象了计算系统的重要领域并利用了自然语言特征,从而简化了程序开发的过程。用脚本编写的程序通常不需要编译(即它们在启动时被解释),但执行速度比从 C 或 Fortran 代码 [61] 编译的等效程序要慢。 Perl(实用提取和报告语言)是一种高级,多范式,解释性脚本语言,由 Larry Wall 于 1987 年创建,作为 GNU 操作系统的补充,以便于解析和报告文本数据 [62]。其核心特征使其成为操纵生物序列数据的理想语言,其在文本格式中得到很好的体现。用 Perl 编写的最早出现的生物信息学软件可以追溯到 1994 年(表 2)。直到 21 世纪后期,由于其极大的灵活性,Perl 无疑是生物信息学的通用语言 [ 63]]。正如拉里·沃尔(Larry Wall)所说,“实现目标的方法不止一种”。BioPerl 于 1996 年的发展(以及 2002 年的首次发布)促成了 Perl 在生物信息学领域的普及 [64]。该 Perl 编程接口提供便于典型但非平凡任务的模块,例如(i)从本地和远程数据库访问序列数据,(ii)在不同文件格式之间切换,(iii)相似性搜索,以及(iv)注释序列数据。 但是,Perl 的灵活性,加上其严格的语法,很容易导致代码可读性低下。这使得 Perl 代码维护变得困难,特别是在几个月或几年后更新软件。与此同时,另一种高级编程语言将成为生物信息学领域的主要参与者。 Python 就像 Perl 一样,是一种高级的多范式编程语言,最初由 Guido van Rossum 于 1989 年开发。Python 更简单的语法使代码读取和维护更容易。2000 年之后,Python 才开始逐渐成为生物信息学中的主要编程语言。除了 Perl 和 Python 之外,一些非脚本编程语言起源于 20 世纪 90 年代早期,后来也加入了生物信息学领域。 首个全基因组测序项目是 1995 年由遗传学家 J. Craig Venter 领导的对流感嗜血杆菌进行的测序,然而正如我们所知道的那样,开始基因组时代的真正转折点是人类基因组在 21 世纪初的正式公布。 人类基因组计划于 1991 年由美国国立卫生研究院(NIH)发起,13 年内耗费 27 亿美元。1998 年,Celera Genomics(一家由 Venter 运营的生物技术公司)领导了一项竞争性私人形式的人类基因组测序组装项目。最终 Celera 支持的该计划用 NIH 项目花费的十分之一成功完成人类基因组进行了测序和组装。两者成本之间的 10 倍差异主要是由于不同的实验策略和 Celera 项目使用了部分 NIH 的数据。 尽管科学界正在经历一段非常激动的时期,但全基因组测序仍需要数百万美元和数年才能完成,甚至对于细菌基因组也是如此。相比之下,如今进行一个人类基因组测序只需花费 1000 美元和不到一周的时间。这种巨大的差异并不令人惊讶,那时即使存在各种文库制备方案,但测序的 reads 仍然要使用 Sanger 毛细管测序仪产生(如下图所示)。最大的测序量也不过是每个 run 产生 96 个长度 800 bp 的 reads,这比二代测序仪要低几个数量级。对人类基因组进行测序(3.0 Gbp)需要大约 40 000 个 runs 才能得到一倍的覆盖率。 除了费力的实验程序外,科学家还必须设计专门的软件来应对这种前所未有的数据量。几个早先基于 Perl 的软件就是在 20 世纪 90 年代中后期开发用于组装全基因组测序 reads 的,比如:PHRAP,Celera Assembler,TIGR Assembler,MIRA,EULER 等等。 基因组学时代的早期,20 世纪 90 年代初出现全球化的信息网络扮演者另一个重要角色。正是通过这个网络,NIH 资助的人类基因组测序项目可以公开其数据。很快,这个网络将在科学界无处不在,特别是在数据和软件共享方面。 20 世纪 90 年代初,Tim Berners-Lee 在欧洲核子研究中心(CERN)担任研究员时发起了万维网,这是一个由相互关联的文件组成的全球信息系统。自 20 世纪 90 年代中期以来,网络已经彻底改变了文化,商业和技术,并在人类历史上第一次实现了近乎即时的通信。 该技术还促成了世界各地许多生物信息学资源库的创建。例如于 1993 年在网上公布的世界上第一个核苷酸序列数据库 EMBL Nucleotide Sequence Data Library。几乎同时,1992 年 GenBank 数据库也成为 NCBI 负责的主要内容之一。然而,那个时候的 GenBank 与今天截然不同,其首次发行是以印刷品和 CD-ROM 的形式。此外,NCBI 于 1994 年开始提供在线服务,随后建立了今天仍在使用的几大主要数据库:Genomes(1995),PubMed(1997)和 Human Genome(1999)。 生物信息学软件通常需要先前了解类 UNIX 操作系统,还需要使用命令行(用于安装和使用)和安装多个软件库(依赖项)才能使用,即使对熟练的生物信息学家而言谈不上直观。但是 Web 资源的兴起扩大并简化了对生物信息学工具的获取,人们可以通过友好的图形用户界面在 Web 服务器上进行操作。越来越多的开发人员尝试通过易于使用的图形 Web 服务器向科学界提供他们的工具,从而无需执行严格的安装过程即可分析数据。核酸研究 杂志每年都会发布关于这些工具的特刊。 蛋白质的第一个三维结构,即肌红蛋白的三维结构,是在 1958 年使用 X 射线衍射实验确定的。然而,Pauling 和 Corey 在 1951 年提出关于蛋白质结构预测并发表了两篇报道 α-螺旋和 β-折叠预测的文章才是第一个真正意义的里程碑。现在,人们可以使用计算机进行计算并预测蛋白质的二级和三级结构。 尽管现代计算机的计算能力不断增加,但对于许多生物分子而言,计算资源仍然是在合理的时间尺度上进行分子动力学模拟的最大困难。好在现在也出现了一些创新,如使用的图形处理单元(GPU)去进行分子动力学模拟,此外,GPU 也开始在需要大量计算能力的其它生物信息学领域发挥作用。 DNA 测序随着二代测序(也称为新一代测序或 NGS)的出现而平民化。这种测序始于 454 焦磷酸测序技术,该技术允许在一台机器对数千至数百万个 DNA 分子进行测序。处理来自 454 数据的黄金标准工具 Newbler 至今仍然由 Roche 维护,直到 2016 年 454 逐步被淘汰。现在其他几家公司和技术正活跃在市场,也有众多工具可用于处理这些数据。 事实上现在有太多的工具以至于很难选择究竟应该用什么。如果这种趋势持续下去,不同团队比较彼此研究结果和重复其他研究组的结果将变得越来越困难。此外,转换到更新和(或)不同的生物信息学工具也都需要额外的培训和测试,这使得研究人员通常不愿放弃他们熟悉的软件。因此必须通过降低计算时间或显着提高质量的结果来证明新工具值得掌握。不过随着公共数据库中存储的生物数据的指数级增加,可用工具的数量增加也相形见绌。 自 2008 年以来,摩尔定律不再是 DNA 测序成本的准确预测因子,因为它们在大规模并行测序技术(https://www.genome.gov/sequencingcosts/)到来后下降了几个数量级。这导致公共数据库(如 GenBank 和 WGS )中的序列呈指数增长(如下图所示),引发了人们对大数据问题的进一步关注。事实上,科学界现在已经产生了超过 exabyte(10 的 18 次方)的数据。 生物信息学项目的繁荣加上数据量呈指数增长需要资助机构逐渐适应。对于绝大多数科学研究,生物信息学项目当然也需要资源。虽然在某些情况下简单的台式机就足够了,但一些项目需要更加强大、昂贵且需要特殊专业知识的基础设施。一些政府资助的专门从事高性能计算的资源已经出现,例如: 高性能计算的重要性也促使一些公司,如亚马逊和微软提供生物信息学服务。 最近与生物信息学相关的一项“进化”是专门研究该领域的研究人员的出现:生物信息学家(bioinformaticians)。不过即使经过 50 多年的生物信息学研究,对什么是生物信息学家仍然没有明确的共识。 例如,一些作者建议将“生物信息学家”这一术语保留给生物信息学领域的专业人士,包括那些开发,维护和部署生物信息学工具的人。另一方面,还有人建议任何生物信息学工具的使用者都应被授予生物信息学家的地位。当然,还有另一个试探性的方法(可能是为了幽默),即通过反面来定义如何不成为一个生物信息学家。 然而可以肯定的是,随着用户友好型工具的显着增加,通常可通过 Galaxy 等综合 Web 服务器获得,以及诸如 SEQanswers 和 BioStar] 等社区的发展。在学术和企业就业市场上,生物信息学家也存在着爆炸性的需求。为了满足这一需要,有必要督促大学调整其生物科学课程的设置。 现在,那些不直接参与生物信息学项目的生命科学家也需要熟练掌握基本概念以了解生物信息学工具的精妙之处,同时避免滥用和错误地结果解读。 国际计算生物学学会根据三个用户类别(生物信息学用户,生物信息学科学家和生物信息学工程师)发布了在其课程中应具备的核心能力指南和建议。所有三个用户类别都包含核心竞争力,例如: 为剩下的两个类别还定义了额外的能力,例如: 在尝试定义生物信息学专业之前,生物信息学这个词本身可能需要适当的定义。事实上计算机的使用已经在生物学以及大多数自然科学(物理学,化学,数学,密码学等)中普遍存在,但有趣的是只有生物学有一个特定的术语来指代计算机的使用。为什么? 首先,生物学在历史上被认为处于“硬”和“软”科学的分界线上。其次,在生物学中使用计算机需要对大分子(即核酸和蛋白质)结构有一定了解。这导致生物学比其他“硬”科学(如物理学和数学)更晚地计算机化。毕竟第一台计算机就是专门用于解决物理领域的数学计算问题。 这些因素的结合可能会解释为什么生物学和计算机之间的联系并不是显而易见的。这也可以解释为什么“生物信息学”一词的使用仍然是常用的。今天,当几乎任何研究工作都需要使用计算机时,人们可能会怀疑这个术语在未来的合理性。生物信息学家 C. Titus Brown 在第 15 届年度生物信息学开源会议上对此进行了有趣的思考,他介绍了当前生物信息学的历史,不过是从 2039 年的一个生物学家角度进行讲述了这一点。在他假设的未来,生物学和生物信息学紧密交织没有必要区分彼此,大家都被称为生物学。 20 世纪后期见证了生物学中计算机的出现,它们的使用以及不断改进的实验室技术使得研究工作日益复杂。尽管对单个蛋白质或基因的测序可能是 20 世纪 90 年代早期的博士论文主题,但博士生现在可以在他/她的研究生阶段就分析许多微生物群落的集体基因组。当时确定蛋白质的一级结构都是复杂的,但是现在可以识别样品的整个蛋白质组。生物学现在已经采用了很多整体方法,但在不同的大分子类别(例如基因组学,蛋白质组学和糖组学)中,每个子学科之间还鲜有交叉。 人们可以预见到下一个飞跃:不是独立研究整个基因组,整个转录组或整个代谢组,而是对整个生物体及其环境进行计算建模,同时考虑所有分子类别。事实上,这一壮举已经在生殖支原体的全细胞模型中实现,其中所有基因,它们的产物和已知代谢相互作用都已在计算机中重建。也许我们很快就会见证一个电子计算机多细胞生物模型。尽管对于数百万到几万亿的细胞建模似乎是不可行的,但必须记住我们现在做的也是十年前在计算能力和技术上认为不可能实现的事情。 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。
起源:1950-1970
生物信息从蛋白质研究开始
第一位生物信息学家

计算机算法的出现
读取 DNA 序列
DNA 序列应用于系统发育推断
生物学和计算机并行发展:1980-1990
靶向和扩增特定基因
使用计算机和专用软件


生物信息学和自由软件运动
台式计算机和新的编程语言
基因组学、结构生物信息学和信息高速路:1990-2000
基因组学时代的黎明

生物信息学在线
超越序列分析:结构生物信息学
高通量生物信息学:2000-2010
二代测序
生物大数据
高性能生物信息学和协作计算
现在和未来的前景:2010 年至今
明确定义生物信息学专业
“生物信息学”一词现在已经过时了吗
将生活整体建模:系统生物学
· 分享链接 https://kaopubear.top/blog/2018-08-15-bioinfohistory/