第一篇 Hi-C 文章: Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome; DOI: 10.1126/science.1181369 TAD 提出: Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions ;doi: 10.1038/nature11082 高分辨率 Hi-C: A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping https://doi.org/10.1016/j.cell.2014.11.021 单细胞 Single cell: Hi-C reveals cell-to-cell variability in chromosome structure doi: 10.1038/nature12593;3D structures of individual mammalian genomes studied by single-cell Hi-C doi:10.1038/nature21429 综述:老师所讲 Hi-C 相关基础知识主要来自于综述 Organization and function of the 3D genome,doi:10.1038/nrg.2016.112 不同分辨率 Hi-C 可以看到的内容不同 5KB 可以看到各种 loop 10KB 可以看到 TAD 50kb 可以看到 TAD 之间的关联 在整个染色体的水平可以看到染色质的位置分布 Cohesin is a protein complex that regulates the separation of sister chromatids during cell division, either mitosis or meiosis. Cohesins hold sister chromatids together after DNA replication until anaphase when removal of cohesin leads to separation of sister chromatids. 转录阻抑物 CTCF CTCF 与靶顺序因子的结合可阻断增强子和启动子的相互作用,从而将增强子的活性限制在一定的功能区域 除了阻断增强子外,CTCF 还可作为染色质屏障阻止异染色质的传播 TargetFinder(Whalen et al. Nat Gen 2016)— an algorithm that uses many functional genomic datasets, including DNase-seq, histone marks, transcription factor (TF) ChIP-seq, gene expression, and DNA methylation data etc. Enhancer–promoter interactions are encoded by complex genomic signatures on looping chromatin ,doi:10.1038/ng.3539 pipeline RIPPLE (Roy et al. NAR 2016) — Also uses functional genomic datasets for feature extraction. 二者共同的发现 Analysis methods for studying the 3D architecture of the genome ,https://doi.org/10.1186/s13059-015-0745-7 流程 定义:A contact map is a matrix with rows and columns representing non-overlapping ‘bins’ across the genome. Each entry in the matrix contains a count of read pairs that connect the corresponding bin pair in a Hi-C experiment. How to determine bin size Two types of approaches to correct bias in the contact map Explicit approach — assuming some known bias Implicit approach — assume no known source of bias and that each locus receives equal sequence coverage after biases are removed 如何进行标准化 HMM(任兵) Arrowhead 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。重要的 Hi-C 相关文献
Chromatin interaction in different resolutions
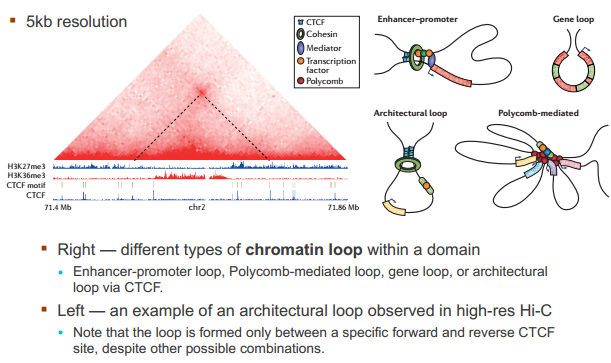
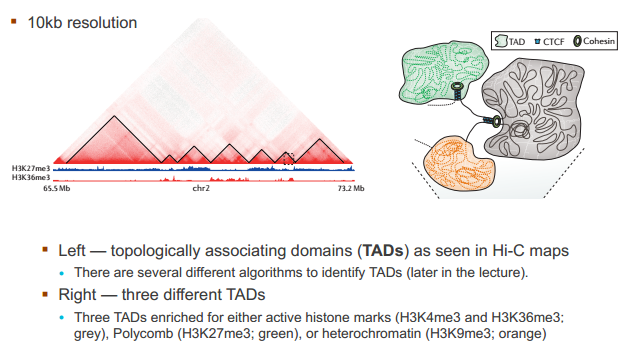
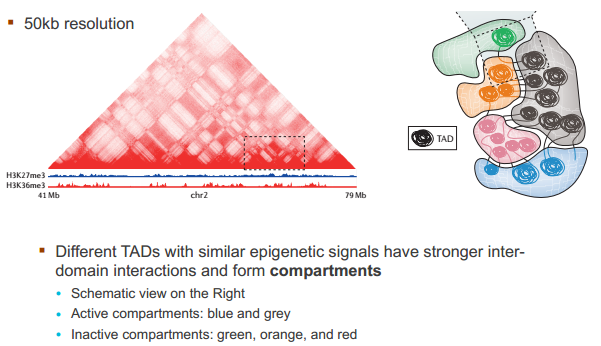
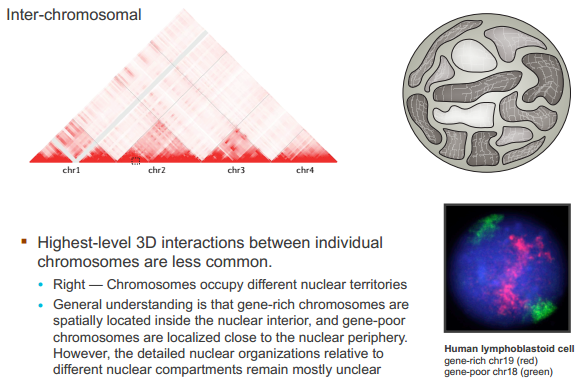
什么造成了所谓的 TAD
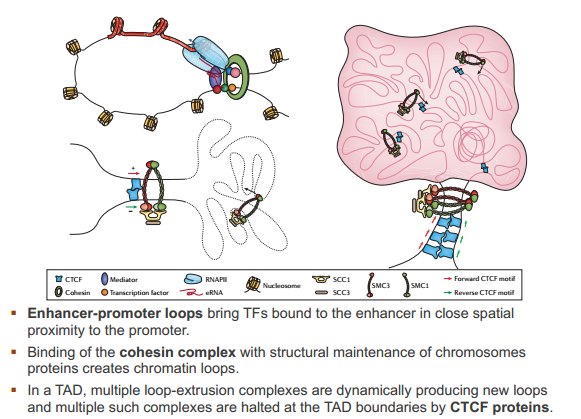
cohesin complex
CTCF proteins
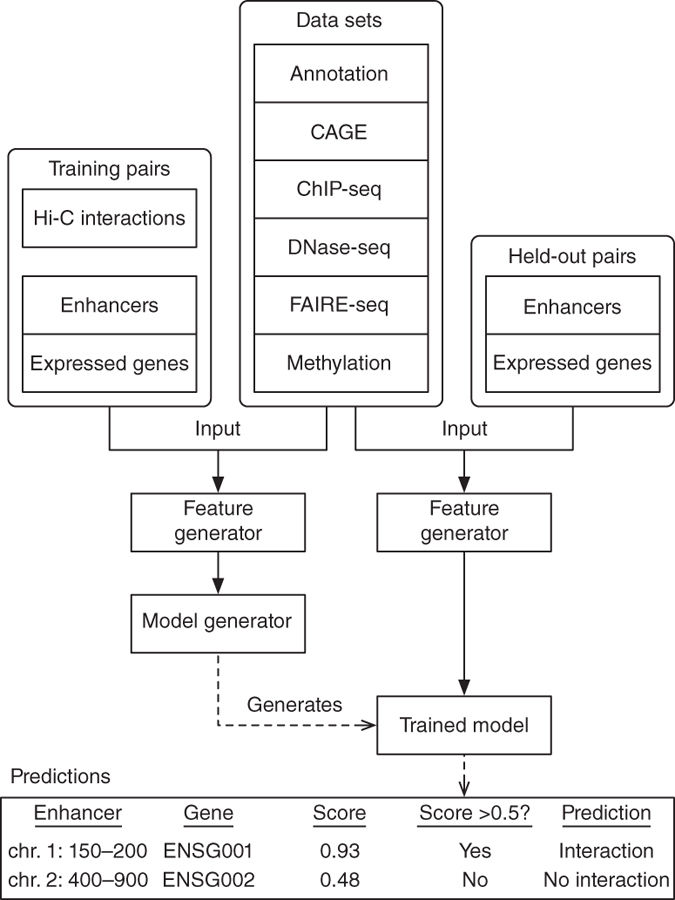
Predicting enhancer-promoter loops 如何预测 EPL
两种类似的算法
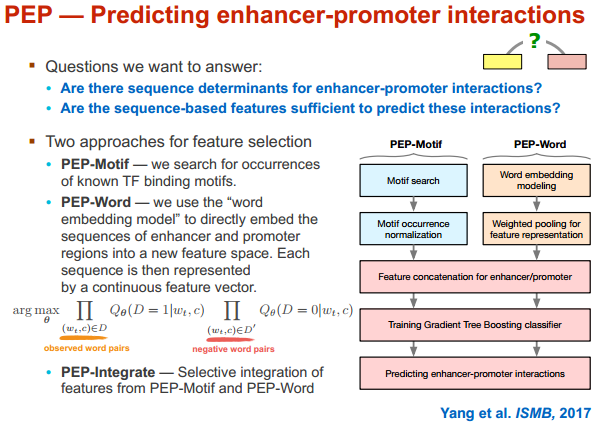
PEP 只用序列信息来进行分析(马坚实验室)
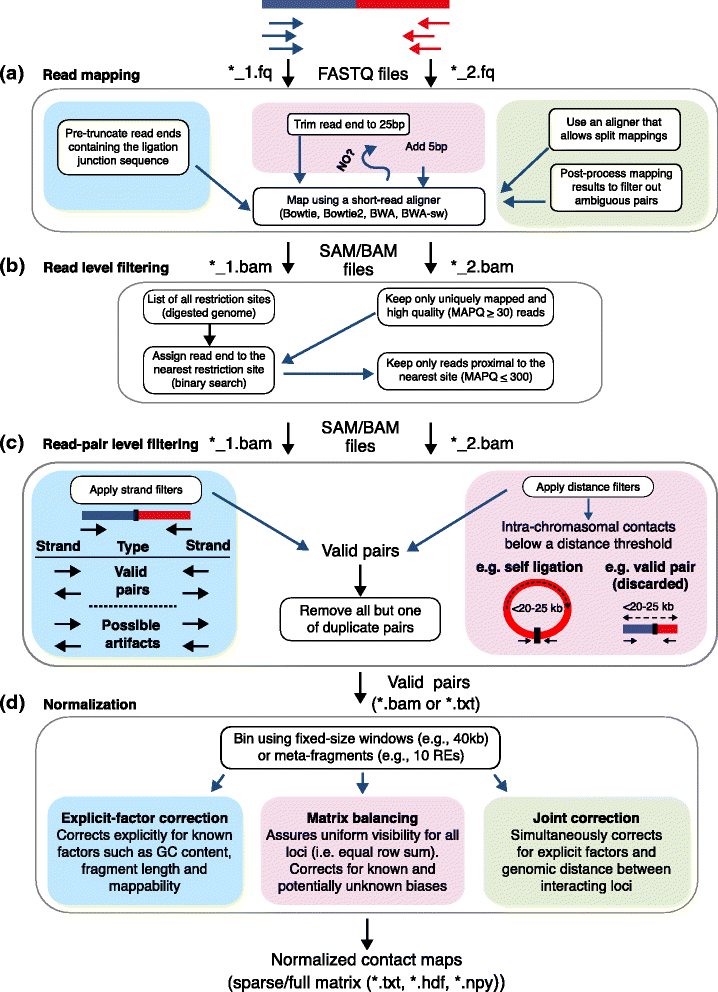
Hi-C 分析流程
contact map
- Restriction enzyme fragment lengths, GC content, and sequence mappability are three major sources of biases in Hi-C data (Yaffe and Tanay, Nat Genet 2011)
- HiCNorm — simpler and faster (Hu et al. Bioinformatics 2012)
- In other words, if there is no bias, the total genome-wide contact summation for each locus will be a constant, i.e., each locus has 'equal visibility'
Contact matrix normalization

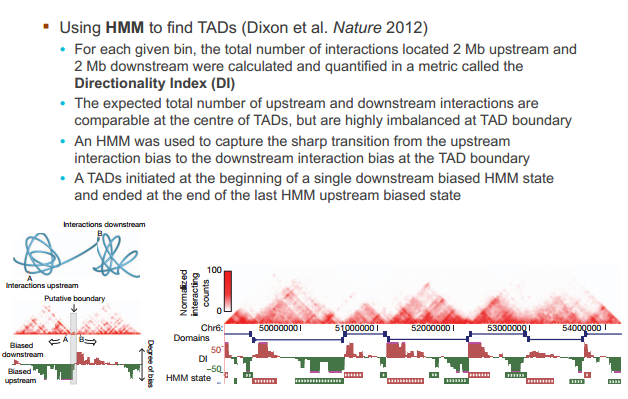
鉴别 TAD 的算法

· 分享链接 https://kaopubear.top/blog/2017-08-11-longxing-bioinfo-hic/