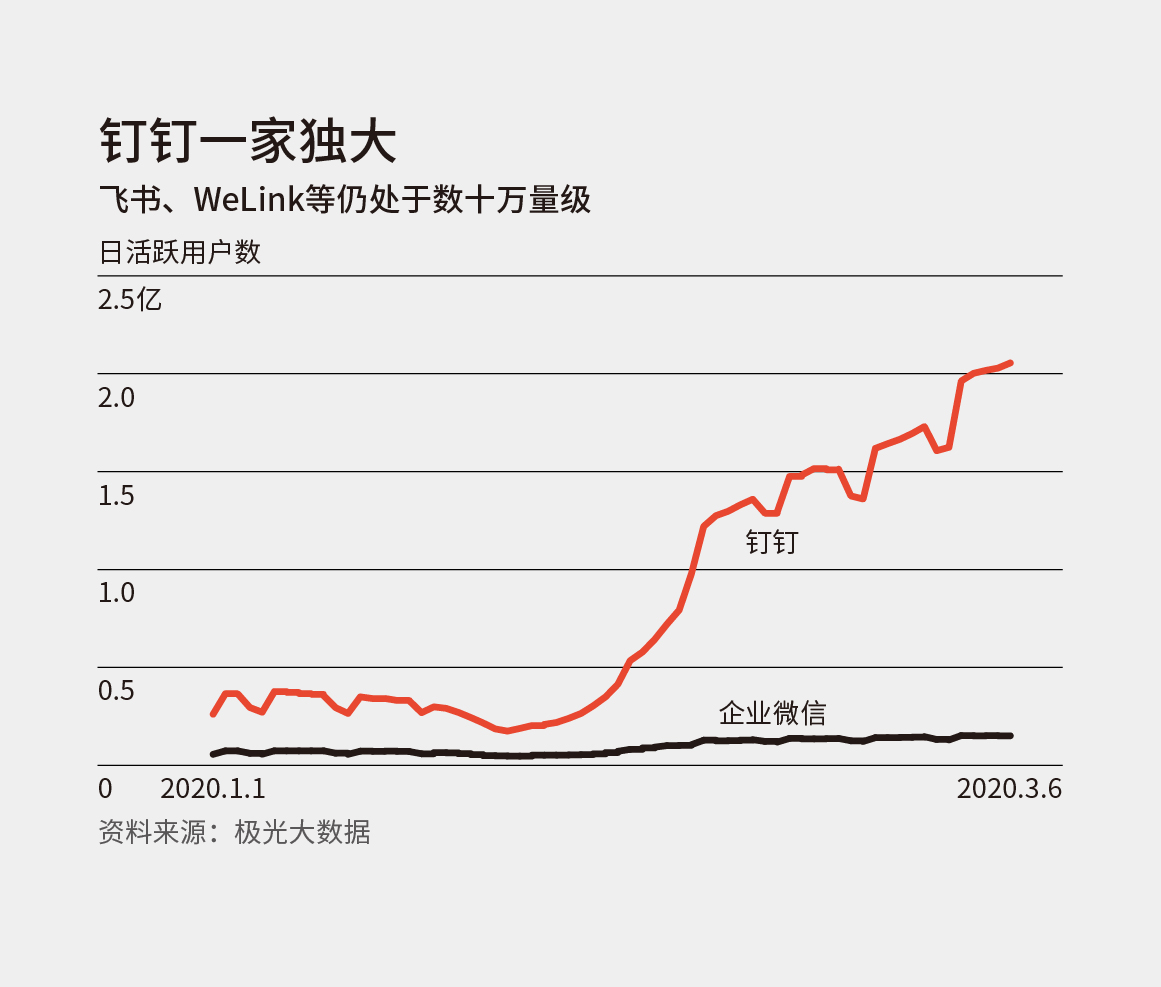
这周的刊首语随便聊聊几个工具。 有传闻,微信即将关闭公众号文章分享到印象笔记的接口。如果过一段时间这个事情成真了,那应该就是所有第三方服务和 app 都要被微信掐死了。 在「财新周刊」最新一期的杂志中,提到了最近几个远程办公工具的一组数据: 今年 1 月初,钉钉的日活用户数已经在 3000 万量级,是企业微信的 5 倍左右,华为和字节跳动则仍在 10 万量级徘徊。据极光大数据,钉钉从 2 月初的数千万日活已上涨超过 2 亿;腾讯会议从 100 多万升至 1000 多万;企业微信也从将近千万升至 1500 万。飞书和 WeLink 则仍在几十万量级。 原来钉钉现在已经到了这样量级,而我日常在用的飞书根本还是排不上号。但是身边接触的人使用飞书的越来越多,以至于我产生了某种程度的认知偏差。 Dimension reduction techniques are widely used to interpret high-dimensional biological data. Features learned from these methods are used to discover both technical artifacts and novel biological phenomena. Such feature discovery is critically import to large single-cell datasets, where lack of a ground truth limits validation and interpretation. Transfer learning (TL) can be used to relate the features learned from one source dataset to a new target dataset to perform biologically-driven validation by evaluating their use in or association with additional sample annotations in that independent target dataset. Zhikai Liang, Yumou Qiu, James C Schnable Abstract Abstract Abstract Abstract 插画素材风格多样、质量超高,网站上提供了 80 个免费的插画,可以免费用于个人和商业项目中。 Astrian 写在 2019 年底的 2010 编年史:我们所爱的流行文化们 最近因为巧合又读了一遍,很多地方都说出了我的感受,但是我并没有能力把过去的 10 年做一个个人化的梳理,因此要再次推荐一下。从一篇文章中能读懂一个人,看到一个人的知识储备。 人们彼此相连,却又彼此成为一座座孤岛。我们无法摆脱孤独,正如我们无法摆脱与他人产生联系。 The greatest designs of modern times What does it take to become a design icon? There‘s more to it than good looks. These 100 products have made our lives simpler, better, and yes, more stylish. 中文报道:http://reader.s-reader.com/article/f5/3892463.html GitHub 和 juypter https://github.com/crazyhottommy/getting-started-with-genomics-tools-and-resources http://rafalab.github.io/pages/teaching.html https://bioinformaticsworkbook.org/ 本周的影音类内容推荐一个私货,「熊言熊语」播客的试播正式上线了。 播客 RSS 地址:https://podcast.kaopubear.top/episodes/feed.xml 「熊言熊语」是一档由 思考问题的熊 主持的播客(podcast)栏目,在双周更新的基础上争取单周更新。目前托管在喜马拉雅,思考问题的熊和他的朋友们一起聊学习聊工作聊生活。 形式 内容 订阅 「熊言熊语」目前托管在喜马拉雅平台,你可以通过苹果播客(Podcast)、Google Podcast、Spotify 以及其他泛用型播客客户端进行订阅收听,也可以在喜马拉雅、网易云音乐和 Bilibili 等国内平台搜索收听,还可以在该播客首页直接收听节目。 https://www.youtube.com/watch?v=F4oUJp76KUY&feature=youtu.be Bob 是一款 Mac 端翻译软件,翻译方式支持划词翻译和截图翻译,翻译引擎支持有道翻译、百度翻译和谷歌翻译。 https://blog.rstudio.com/2020/03/17/rstudio-1-3-the-little-things/ https://www.jiqizhixin.com/articles/2020-03-18-10 介绍三个比较不错的在线写作网站以及他们的特点和问题 访问和使用 Telegraph 不需要注册账号与下载软件,只需在网页浏览器中访问 http://telegra.ph/ 便可看到简约的界面,填入要发布的内容即可匿名发布。内容发布之后,只要清除浏览器的缓存,便无法再编辑文章。内容发布之后,不能追溯到文章作者和发布者。类似于一个匿名博客,问题是在国内无法正常访问。 WTDF.io 与 Telegraph 在编辑体验上类似,编辑器本身提供了字号、字体和背景配色,国内环境可以正常访问,支持富文本导出,分享时也能保证风格和排版不会出现太大的差异。wtdf 同时支持 markdown 和富文本,和 typora 类似。 一个台湾工程师开发的即时在线协作 markdown 编辑器,在使用上是一个相对传统的 markdown 编辑器,协作的体验非常好。 最近三年 Notion 越来越火了,尤其是过去一年在国内获得了不少关注,其实和它使用体验类似甚至某些层面还更优的并不止一款工具,Coda 就是一个。最近有 一篇文章 介绍了 Coda 的发展历史。 现在大多数录屏直播或者视频都没有分享者的脸,其实这个问题在游戏直播界很早就有推荐的东西了,也有不错的视频会议工具可以使用,比如 zoom 和 loom。 关于 OBS 的使用介绍和教程 https://obsproject.com/wiki/ 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。刊首语
传闻微信再次收紧第三方接入服务
很多国内的效率类笔记类工具一开始主打功能都包括和微信的整合,比如滴答清单比如印象笔记这类。
之前写分享和教程都会特意留意一下能不能和微信联动一下,最早印象笔记可以存微信群聊天记录,滴答清单可以直接发送任务。后来这些一个一个都没了。
不止一次收到类似于「与其用这用那不如好好研究下微信怎么用」的评论,我研究啥啊,研究私域流量,研究局域网么?
微信是有提醒功能也能聊天消息收藏转笔记,怎么着,这就是未来的 All in One 工具么 🙃。从移动办公工具看认知偏差
专业文献
projectR: An R/Bioconductor package for transfer learning via PCA, NMF, correlation, and clustering.
RESULTS:
We developed an R/Bioconductor package, projectR, to perform TL for analyses of genomics data via TL of clustering, correlation, and factorization methods. We then demonstrate the utility TL for integrated data analysis with an example for spatial single-cell analysis.
AVAILABILITY:
projectR is available on Bioconductor and at https://github.com/genesofeve/projectRGenome-phenome wide association in maize and Arabidopsis identifies a common molecular and evolutionary signature
Molecular Plant 2020 March 11
Linking natural genetic variation to trait variation can help determine the functional roles of different genes play. Often variation of one or several traits are assessed separately. High throughput phenotyping and data mining can capture dozens or hundreds of traits from the same individuals. Here we test the association between markers within a gene and many traits simultaneously. This Genome-Phenome Wide Association Study (GPWAS) is both a multi-marker and multi-trait test. Genes identified using GPWAS with 260 phenotypic traits in maize were enriched for genes independently linked to phenotypic variation. Traits associated with classical mutants were consistent with reported phenotypes for mutant alleles. Genes linked to phenomic variation in maize using GPWAS shared molecular, population genetic, and evolutionary features with classical mutants in maize. Genes linked to phenomic variation in Arabidopsis using GPWAS are significantly enriched in genes with known loss of function phenotypes. GPWAS may be an effective strategy to identify genes where loss of function alleles will produce mutant phenotypes. The shared signatures present in classical mutants and genes identified using GPWAS may be markers for genes with a role in specifying plant phenotypes generally, or pleiotropy specifically.RASflow: an RNA-Seq analysis workflow with Snakemake.
BACKGROUND:
With the cost of DNA sequencing decreasing, increasing amounts of RNA-Seq data are being generated giving novel insight into gene expression and regulation. Prior to analysis of gene expression, the RNA-Seq data has to be processed through a number of steps resulting in a quantification of expression of each gene/transcript in each of the analyzed samples. A number of workflows are available to help researchers perform these steps on their own data, or on public data to take advantage of novel software or reference data in data re-analysis. However, many of the existing workflows are limited to specific types of studies. We therefore aimed to develop a maximally general workflow, applicable to a wide range of data and analysis approaches and at the same time support research on both model and non-model organisms. Furthermore, we aimed to make the workflow usable also for users with limited programming skills.
RESULTS:
Utilizing the workflow management system Snakemake and the package management system Conda, we have developed a modular, flexible and user-friendly RNA-Seq analysis workflow: RNA-Seq Analysis Snakemake Workflow (RASflow). Utilizing Snakemake and Conda alleviates challenges with library dependencies and version conflicts and also supports reproducibility. To be applicable for a wide variety of applications, RASflow supports the mapping of reads to both genomic and transcriptomic assemblies. RASflow has a broad range of potential users: it can be applied by researchers interested in any organism and since it requires no programming skills, it can be used by researchers with different backgrounds. The source code of RASflow is available on GitHub: https://github.com/zhxiaokang/RASflow.
CONCLUSIONS:
RASflow is a simple and reliable RNA-Seq analysis workflow covering many use cases.
KEYWORDS:
RNA-Seq; Snakemake; Workflow
PMID: 32183729 DOI: 10.1186/s12859-020-3433-xBenchmarking of computational error-correction methods for next-generation sequencing data
BACKGROUND:
Recent advancements in next-generation sequencing have rapidly improved our ability to study genomic material at an unprecedented scale. Despite substantial improvements in sequencing technologies, errors present in the data still risk confounding downstream analysis and limiting the applicability of sequencing technologies in clinical tools. Computational error correction promises to eliminate sequencing errors, but the relative accuracy of error correction algorithms remains unknown.
RESULTS:
In this paper, we evaluate the ability of error correction algorithms to fix errors across different types of datasets that contain various levels of heterogeneity. We highlight the advantages and limitations of computational error correction techniques across different domains of biology, including immunogenomics and virology. To demonstrate the efficacy of our technique, we apply the UMI-based high-fidelity sequencing protocol to eliminate sequencing errors from both simulated data and the raw reads. We then perform a realistic evaluation of error-correction methods.
CONCLUSIONS:
In terms of accuracy, we find that method performance varies substantially across different types of datasets with no single method performing best on all types of examined data. Finally, we also identify the techniques that offer a good balance between precision and sensitivity.
PMID: 32183840
DOI: 10.1186/s13059-020-01988-3BANDITS: Bayesian differential splicing accounting for sample-to-sample variability and mapping uncertainty.
Alternative splicing is a biological process during gene expression that allows a single gene to code for multiple proteins. However, splicing patterns can be altered in some conditions or diseases. Here, we present BANDITS, a R/Bioconductor package to perform differential splicing, at both gene and transcript level, based on RNA-seq data. BANDITS uses a Bayesian hierarchical structure to explicitly model the variability between samples and treats the transcript allocation of reads as latent variables. We perform an extensive benchmark across both simulated and experimental RNA-seq datasets, where BANDITS has extremely favourable performance with respect to the competitors considered.
PMID: 32178699
DOI: 10.1186/s13059-020-01967-8Transcriptional regulation of genes bearing intronic heterochromatin in the rice genome.
Intronic regions of eukaryotic genomes accumulate many Transposable Elements (TEs). Intronic TEs often trigger the formation of transcriptionally repressive heterochromatin, even within transcription-permissive chromatin environments. Although TE-bearing introns are widely observed in eukaryotic genomes, their epigenetic states, impacts on gene regulation and function, and their contributions to genetic diversity and evolution, remain poorly understood. In this study, we investigated the genome-wide distribution of intronic TEs and their epigenetic states in the Oryza sativa genome, where TEs comprise 35% of the genome. We found that over 10% of rice genes contain intronic heterochromatin, most of which are associated with TEs and repetitive sequences. These heterochromatic introns are longer and highly enriched in promoter-proximal positions. On the other hand, introns also accumulate hypomethylated short TEs. Genes with heterochromatic introns are implicated in various biological functions. Transcription of genes bearing intronic heterochromatin is regulated by an epigenetic mechanism involving the conserved factor OsIBM2, mutation of which results in severe developmental and reproductive defects. Furthermore, we found that heterochromatic introns evolve rapidly compared to non-heterochromatic introns. Our study demonstrates that heterochromatin is a common epigenetic feature associated with actively transcribed genes in the rice genome.
PMID: 32187179 DOI: 10.1371/journal.pgen.1008637数据库
免费可商用插画素材
https://lab.streamlineicons.com/
好书好文
2010 编年史:我们所爱的流行文化们
在文章中他写到这样一段话:
现代百大设计最佳产品。
早在 1959 年,美国《财富》杂志就启动了旨在发掘现代 100 种设计最佳产品的行动。该榜单由伊利诺伊理工学院设计研究所所长杰伊·多布林(Jay Doblin)编制,基于对那个时代 100 名顶尖设计师、建筑师和设计教师的调查完成。其结果完美地说明了上世纪中期的设计理念,同时也反映了当代的品味,上榜产品包括保时捷的时尚跑车,甚至还有伊姆斯(Eames)、阿尔托(Alalto)和萨里宁(Saarinen)的更时尚的扶手椅等。
2019 年,为了纪念最初榜单发布 60 周年,《财富》杂志与现由丹尼斯·韦尔(Denis Weil)担任院长的 IIT 设计学院合作,重新创建了这项调查。研究人员尽可能地遵循多布林的方法,并进行了明智的现代调整,就他们认为真正伟大的发明对教育家、有影响力的人、自由设计师和公司设计团队进行民意调查。经过一年多的策划、调查和整理,为我们呈现了“现代百大设计最佳”产品。
https://fortune.com/longform/100-best-designs/
学习素材
远程学习怎么学
https://jupyter4edu.github.io/jupyter-edu-book/
http://tljh.jupyter.org/en/latest/
https://github.blog/2020-03-18-set-up-your-digital-classroom-with-github-classroom/
Resources and tips for teaching (with) R remotelyUnix, R and python tools for genomics and data science
Teaching resources
Bioinformatics Workbook
https://github.com/ISUgenomics/bioinformatics-workbook影音推荐
熊言熊语

Creating an R data package (cord19) in RStudio
介绍如何创建一个 R 的数据包。工具
Mac 端翻译软件 Bob
RStudio 1.3 Preview: The Little Things
RStudio 发布了新的预览版本,有几个比价不错的更新,其中比较重要的两个如下:
GitHub 移动 App 正式上线
据官方介绍,移动 app 有四个主要的特性功能值得关注。有了这些功能,用户可以在手机上浏览代码、管理工作日程和任务、评价其他工作和项目、合并和管理分支等。优秀的网页 Markdown 写作工具
Coda 一款主打 No Code 的工具
Coda 的核心思想是:在线文档像一块画布,在里面可以添加按钮以及各种控件,文档就变得像一个 app。他们 2014 年开始做,闭门造车做了 3 年才上线;在上线前就融资 $6 千万,估值 $4 个亿。
Coda 同 Airtable、Notion 等工具主导思想都是 No-Code,我们曾经习惯的笔记终将想数据库的形式迈进,在 Coda 看来,未来的软件本质就是文档,或者说在文档里完全可以呈现出一个 APP。
关于 No-code,少数派作者整理了一个Notion 网页有不少信息资料可以参考学习。关于 Coda 的介绍也可以参考少数派中的几篇教程。
共享屏幕且露脸
不过这里要推荐的工具是OBS,OBS 是一个全平台且开源的直播和录播工具,非常强大,可以叠加多个画面,捕捉窗口和屏幕都不是问题。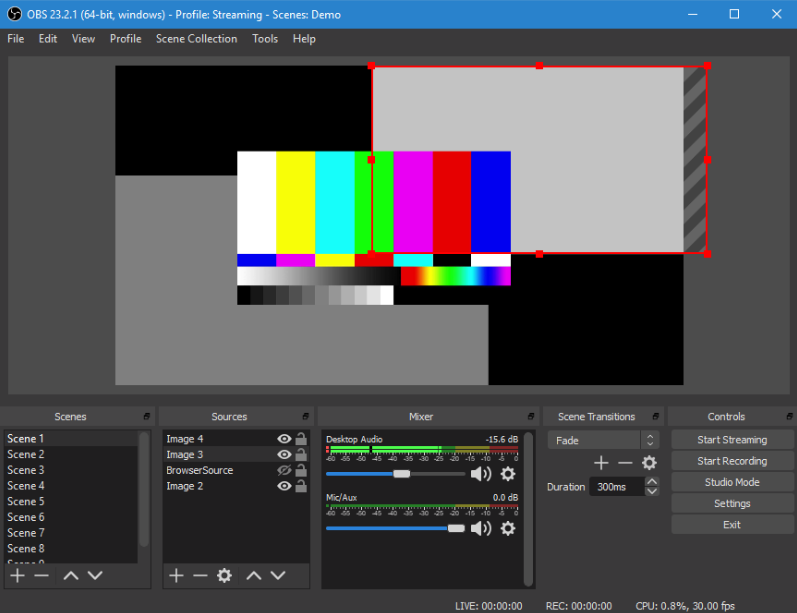
· 分享链接 https://kaopubear.top/blog/2020-03-22-weeklyshare4/