本周把自己一年多没更新的个人公众号(思考问题的熊)更新了一次,结果很快就有了五十多条留言。 不知道你有没有一个自己很久没有留意却突然发现还有人在关注的东西,不妨分享一下。 Circular RNAs (circRNAs) are a unique class of RNA molecule identified more than 40 years ago which are produced by a covalent linkage via back-splicing of linear RNA. Recent advances in sequencing technologies and bioinformatics tools have led directly to an ever-expanding field of types and biological functions of circRNAs. In parallel with technological developments, practical applications of circRNAs have arisen including their utilization as biomarkers of human disease. Currently, circRNA-associated bioinformatics tools can support projects including circRNA annotation, circRNA identification and network analysis of competing endogenous RNA (ceRNA). In this review, we collected about 100 circRNA-associated bioinformatics tools and summarized their current attributes and capabilities. We also performed network analysis and text mining on circRNA tool publications in order to reveal trends in their ongoing development.** circRNA 发展历史 Experiments that profile nascent RNA are growing in popularity; however, there is no standard analysis pipeline to uniformly process the data and assess quality. Here, we introduce PEPPRO, a comprehensive, scalable workflow for GRO-seq, PRO-seq, and ChRO-seq data. PEPPRO produces uniform processed output files for downstream analysis, including alignment files, signal tracks, and count matrices. Furthermore, PEPPRO simplifies downstream analysis by using a standard project definition format which can be read using metadata APIs in R and Python. For quality control, PEPPRO provides several novel statistics and plots, including assessments of adapter abundance, RNA integrity, library complexity, nascent RNA purity, and run-on efficiency. PEPPRO is restartable and fault-tolerant, records copious logs, and provides a web-based project report for navigating results. It can be run on local hardware or using any cluster resource manager, using either native software or our provided modular Linux container environment. PEPPRO is thus a robust and portable first step for genomic nascent RNA analysis. MicroRNAs (miRNAs) are short, non-coding RNAs that modulate the translation-rate of messenger RNAs (mRNAs) by directing the RNA-induced silencing complex to sequence-specific targets. In plants, this typically results in cleavage and subsequent degradation of the mRNA. Degradome sequencing is a high-throughput technique developed to capture cleaved mRNA fragments and thus can be used to support miRNA target prediction. The current criteria used for miRNA target prediction were inferred on a limited number of experimentally validated A. thaliana interactions and were adapted to fit these specific interactions; thus, these fixed criteria may not be optimal across all datasets (organisms, tissues or treatments). We present a new tool, PAREameters, for inferring targeting criteria from small RNA and degradome sequencing datasets. We evaluate its performance using a more extensive set of experimentally validated interactions in multiple A. thaliana datasets. We also perform comprehensive analyses to highlight and quantify the differences between subsets of miRNA–mRNA interactions in model and non-model organisms. Our results show increased sensitivity in A. thaliana when using the PAREameters inferred criteria and that using data-driven criteria enables the identification of additional interactions that further our understanding of the RNA silencing pathway in both model and non-model organisms. https://benubah.github.io/r-community-explorer/rugs.html 该页面总结了 R 语言近年来社区的发展情况,女性用户的分析以及历届 Google Summer of Code 中 R 相关的项目。 最近有 一篇文章 从三个纬度介绍了 R 这 20 年的发展。 Keith McNulty 总结了可以节省时间的10 个 R 操作技巧 9to5mac 发布了一篇关于的教程,介绍了Mac 触控板 ForceTouch 可以做什么,包括如何设置 ForceTouch 以及哪些应用可以使用 ForceTouch。 关于如何定量 lncRNA,19 年有一篇发表在 Gigascience 的文章进行了不同方法的测试,整体的结论如下: Pseudoalignment methods and RSEM detect more lncRNAs and correlate highly with simulated ground truth. On the contrary, HTSeq and featureCounts often underestimate lncRNA expression. Antisense lncRNAs are poorly quantified by alignment-based gene quantification methods, which can be improved using stranded protocols and pseudoalignment methods. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6897288/ 伪比对方法和 RSEM 检测到更多的 lncRNAs,反义 lncRNAs 通过基于比对的基因量化方法难以量化。关于这个问题在 GitHub 有一个简单的讨论:https://github.com/alexdobin/STAR/issues/848 https://bioinformaticsdotca.github.io/ Erik Gahner Larsen 整理了目前可以找到的 ggplot2 相关教程合集 awesome-ggplot2 Hadley Wickham 主编的第三版 ggplot2 教程已经在路上了,服气。 这个简短的视频说明了 RNAseq 是如何工作的,以及如何研究中使用它。Jose Manuel Garcia Manteiga 解释了如何有效地设计 RNAseq 实验以及可以从这项技术中获得的结果。 十分钟的视频把转录组分析相关内容的方方面面今本都涉及到了,很喜欢这样的讲课方式。如果有机会做一些类似的小视频就好了,不过机会在哪呢 ╮( ̄ ▽  ̄)╭ https://www.datawrapper.de/ 如何使用 datawrapper https://academy.datawrapper.de/ 和语雀出自同一家,AntV 是蚂蚁金服全新一代数据可视化解决方案,致力于提供一套简单方便、专业可靠、无限可能的数据可视化最佳实践。近日其 G2 正式发布了 4.0 版本。 微信这个让人又恨又不得不用的东西,有时候不仅不得不用,可能一次还得用两个。这里就涉及到了微信双开的问题。在 android 系统上,现在多数国产厂商都适配了微信双开。至于 Windows 和 macOS 其实也都有自己的方法(其实都是命令行启动),供各位参考。 Windows 上的使用方式 macOS 本身是利用 nohup 命令让微信进行后台运行,如下所示,原则上,这种方法可以同时开启 N 个微信。 该周刊每周六发布,暂时作为「素材分享学习小组」内部资料,我们会挑选部分内容进行公开分享。 如果想要加入「素材分享学习小组」可以参考申请说明,如果想和我们交流,欢迎在评论区留言,我们下周见! 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。刊首语
很多人都在反馈自己过去三四年发生和经历的事情,很神奇的感受,看似没有交集但是却也没有断了联系。
专业文献
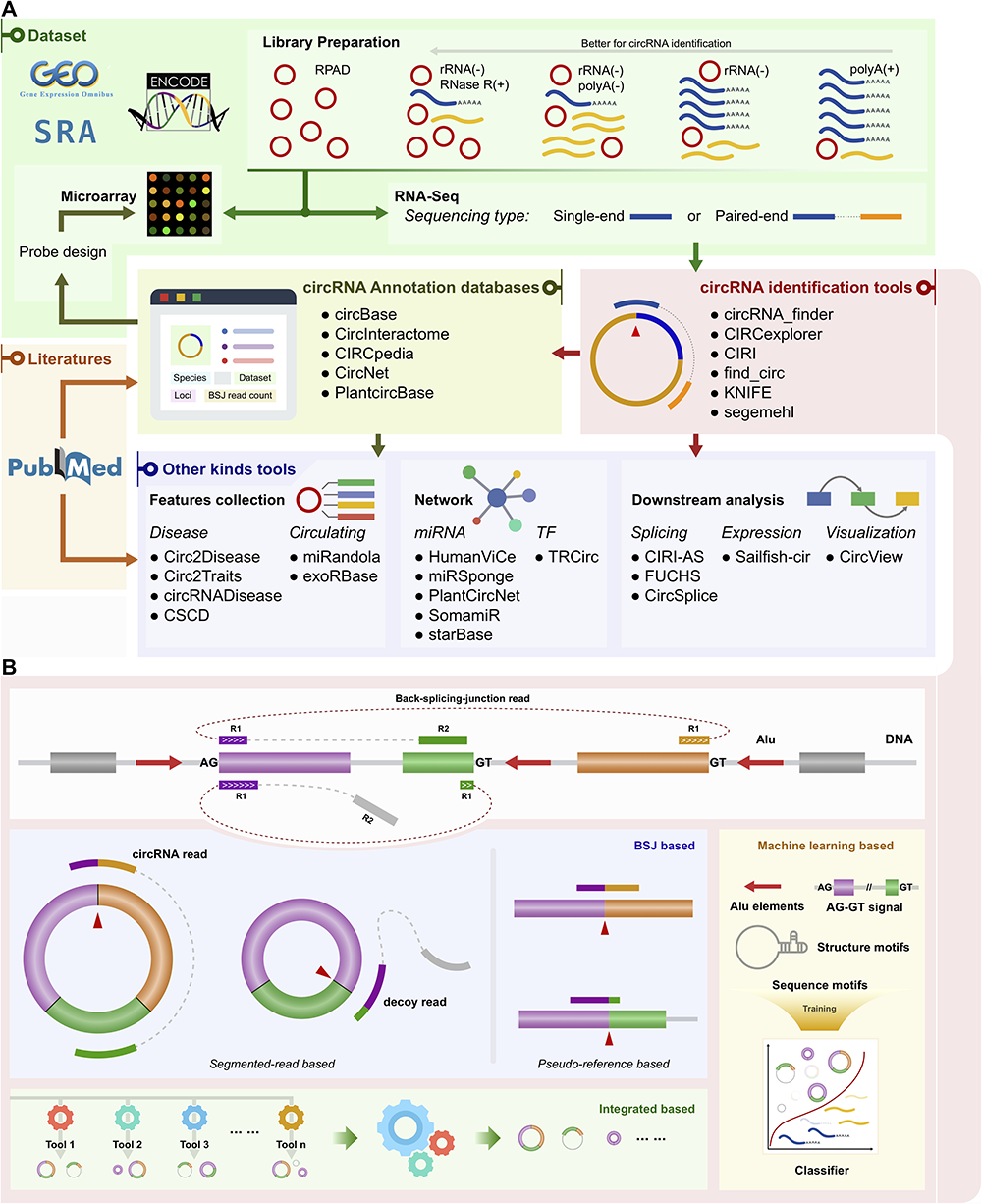
The Bioinformatics Toolbox for circRNA Discovery and Analysis
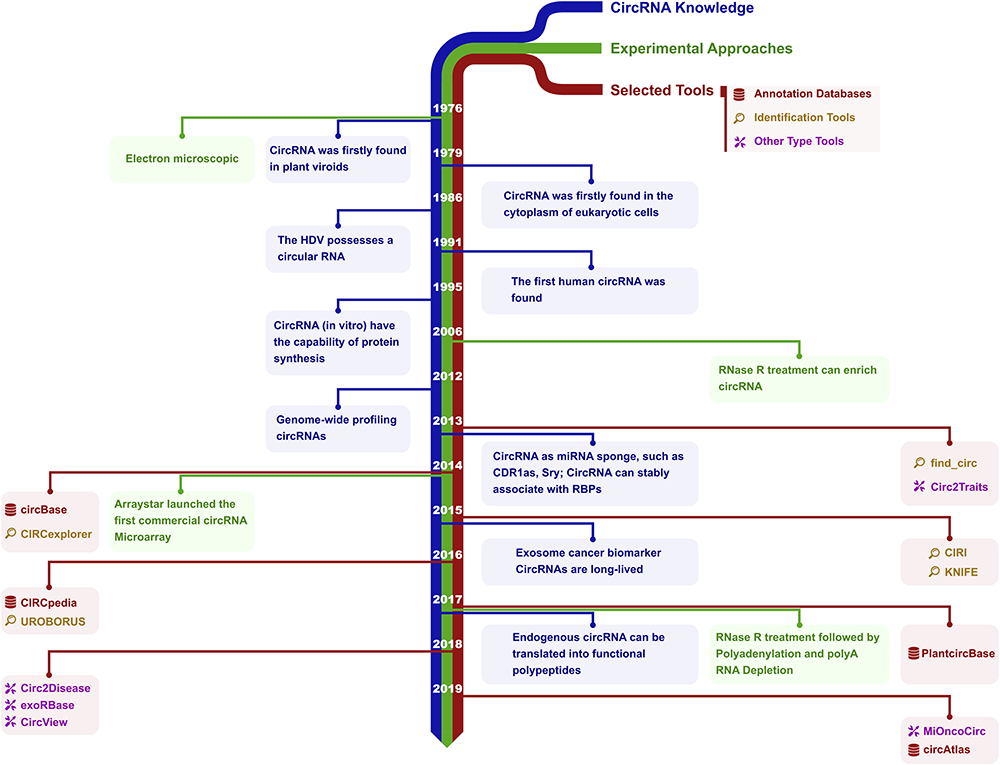
circRNA 分析工具
Quality control and processing of nascent RNA profiling data
Publisher URL: http://biorxiv.org/cgi/content/short/2020.02.27.956110v1
DOI: https://doi.org/10.1101/2020.02.27.956110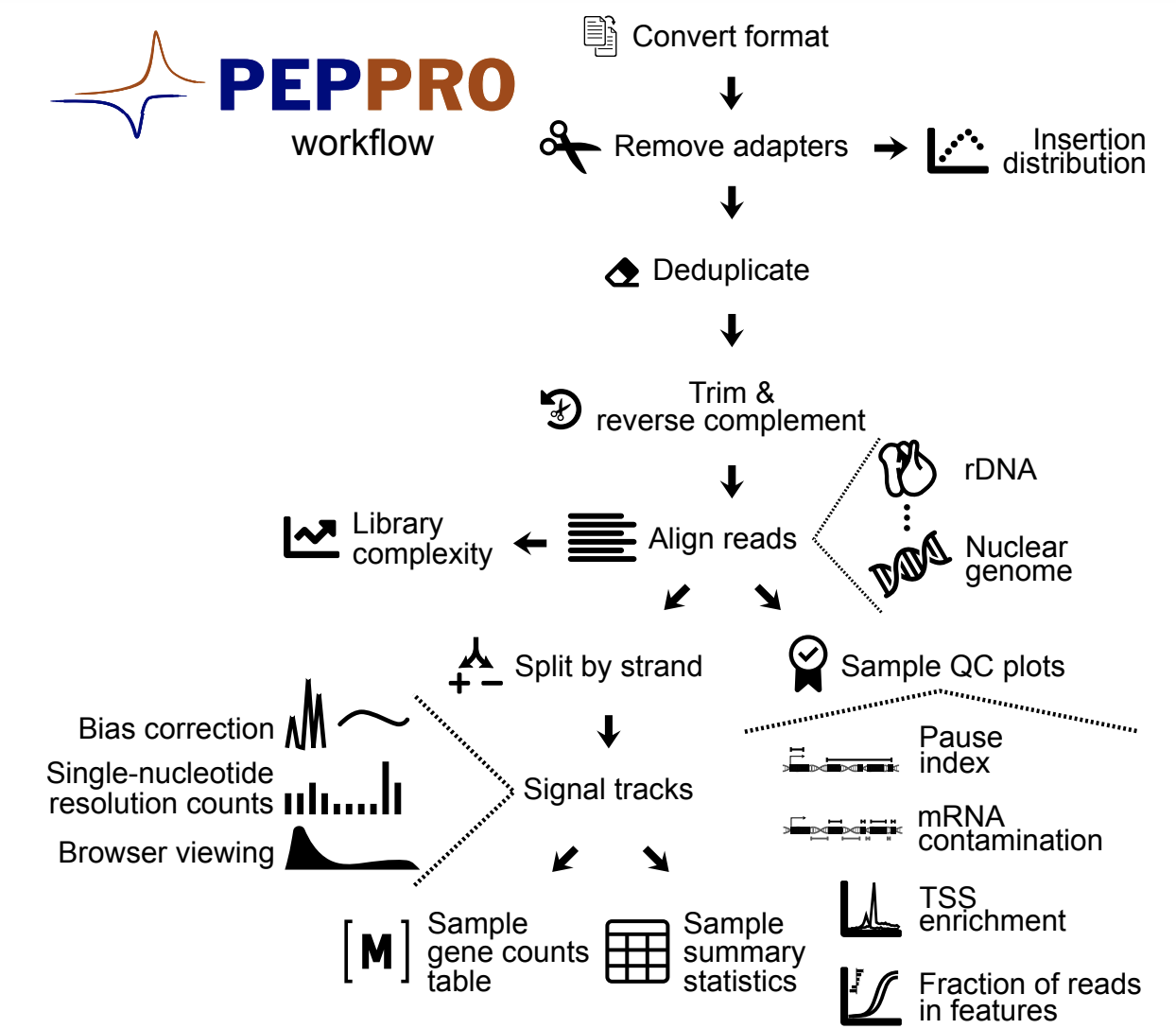
PAREameters: a tool for computational inference of plant miRNA–mRNA targeting rules using small RNA and degradome sequencing data
Publisher URL: https://academic.oup.com/nar/article/48/5/2258/5707202
DOI: https://doi.org/10.1093/nar/gkz1234
数据库
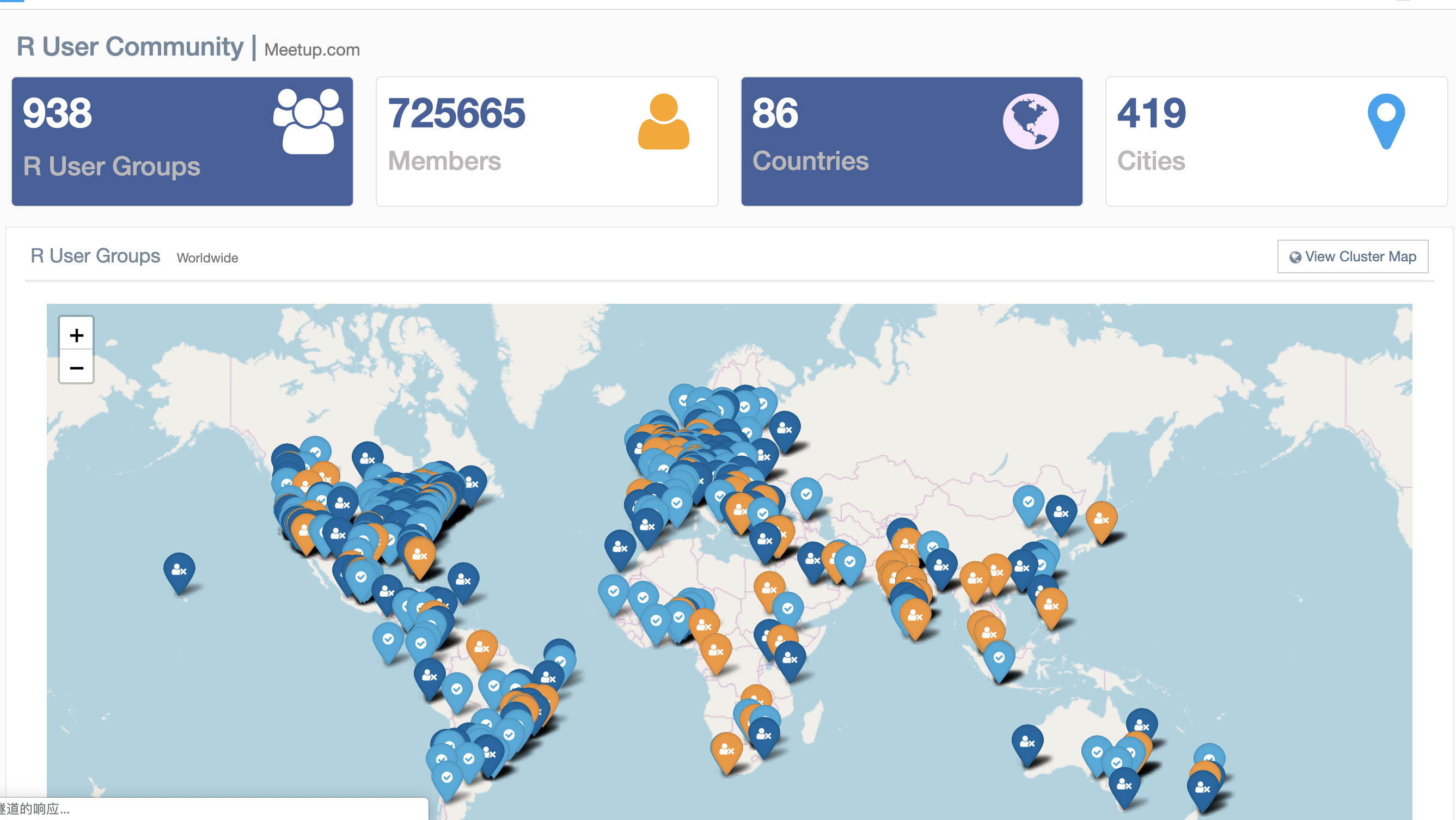
R 社区的发展探索
好书好文
R 的 20 年
10 个节省时间的 R 操作技巧
patchwork packageRenv. to keep pipingMac 触控板 ForceTouch 可以做什么
学习素材
lncRNA 定量注意事项
Canadian Bioinformatics Workshops
Canadian Bioinformatics Workshops 旗下的生物信息教程合集基本上所有内容都可以公开获取
ggplot2 教程合集
各个角度的教程都有,可以学习。ggplot2: Elegant Graphics for Data Analysis
影音推荐
理解 RNAseq 是如何工作的

(我从油管做了一个搬运放到了 B 站,嵌入了英文字幕)工具
Datawrapper
Datawrapper 是一种简单的可视化工具,无需编程基础。大量的媒体和记者都在使用这个工具进行数据的可视化,官方也提供了详细的教程。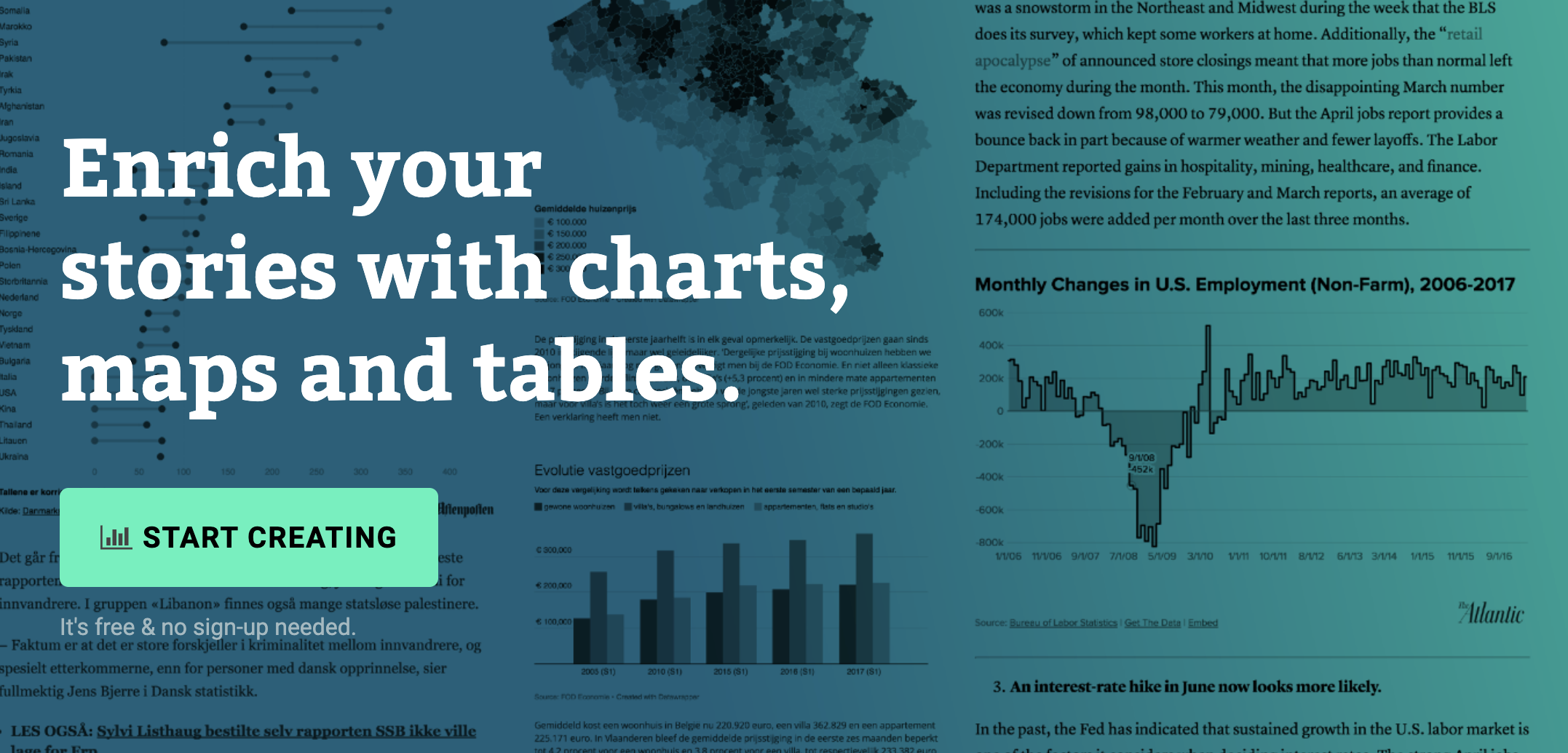
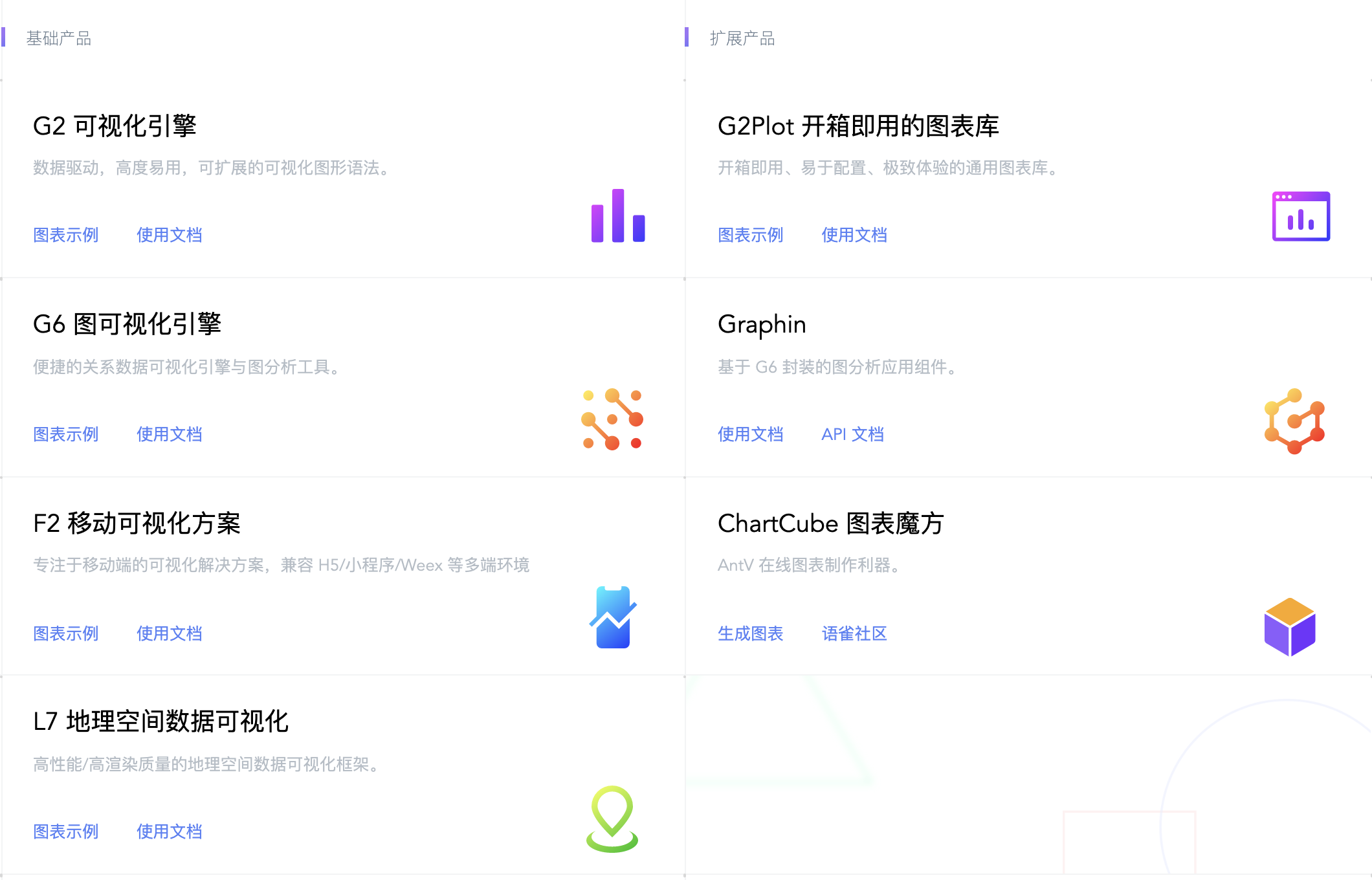
AntV
技巧
微信双开
就是以极快的速度连续点击微信图标有两种思路,我比较喜欢的是在微信安装目录新建一个 bat 文件,里面写两行 start WeChat.ext 。在把这个文件做一个快捷方式到桌面。也可以直接写绝对路径,把文件直接放到桌面。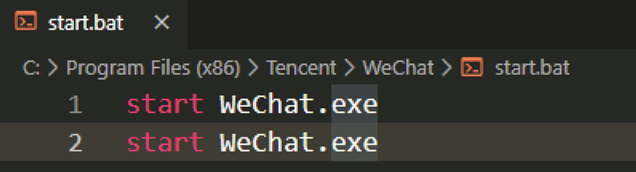
nohup /Applications/WeChat.app/Contents/MacOS/WeChat > /dev/null 2>&1 &
订阅
完成前期测试运行后将择期通过博客和公众号等形式同步更新,届时将发布具体的订阅形式。交流
· 分享链接 https://kaopubear.top/blog/2020-03-06-weeklyshare2/