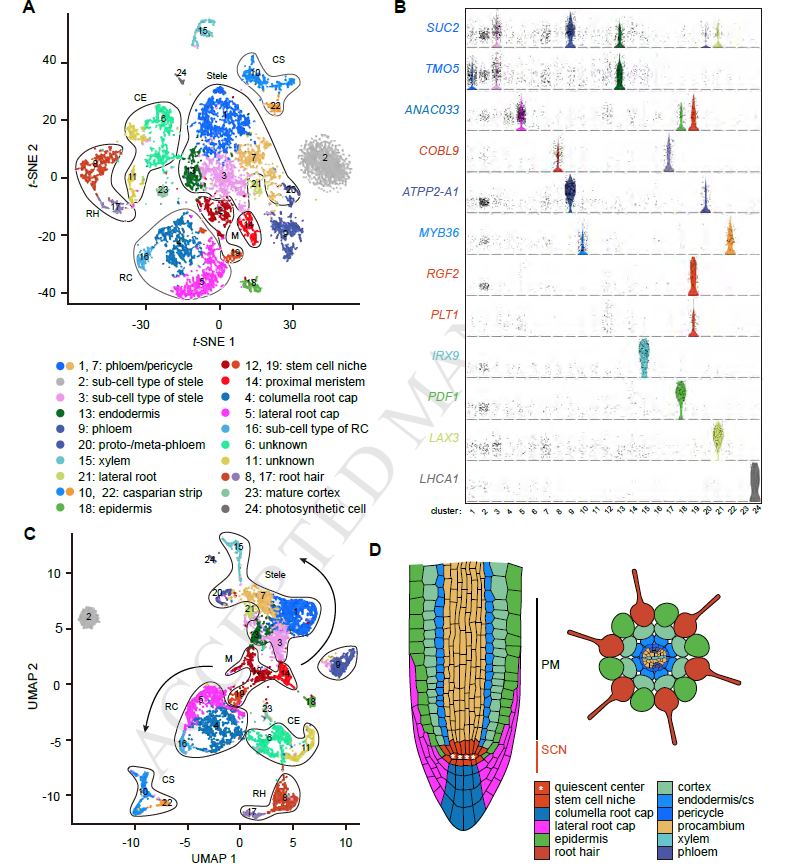
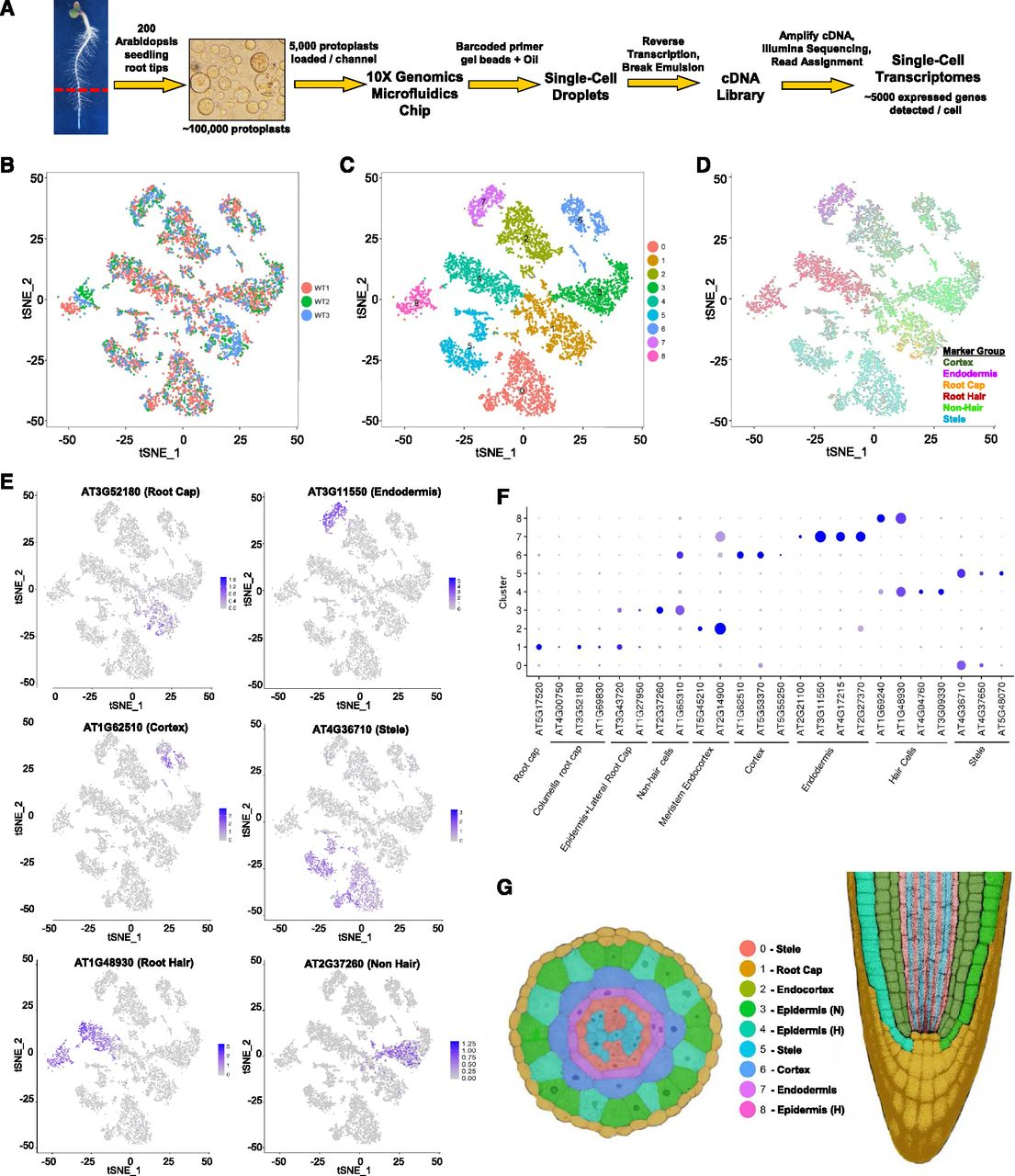
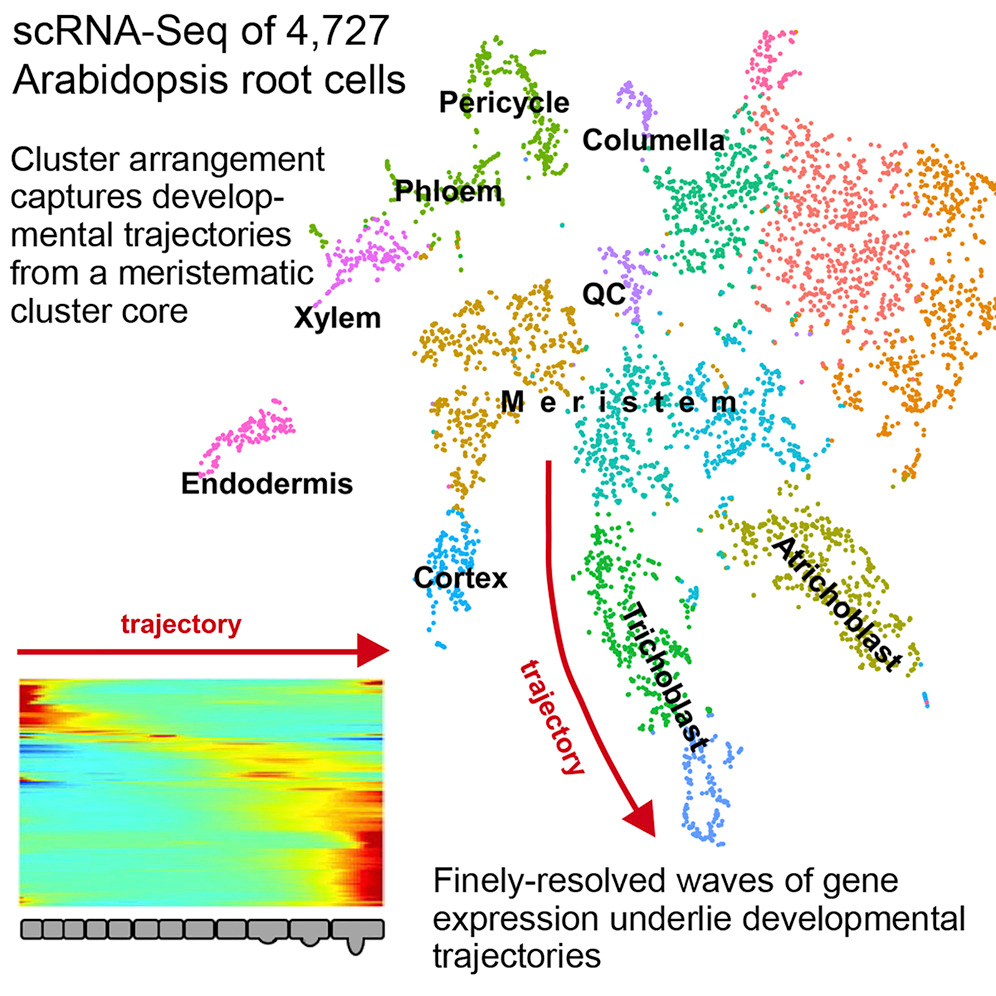
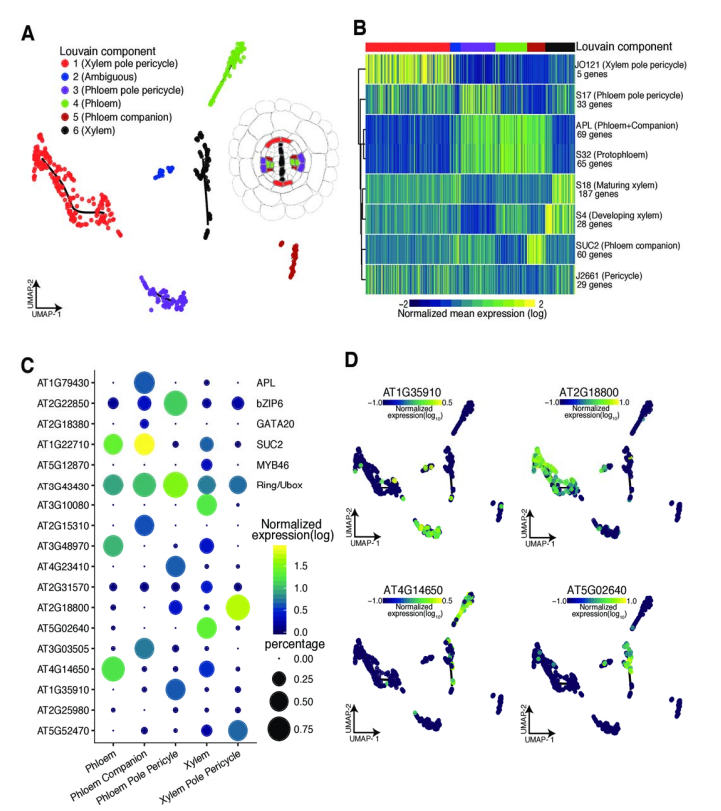
DOI(url): https://doi.org/10.1016/j.molp.2019.04.004 发表日期:April 17, 2019 国内首篇植物相关单细胞文章,两个一作还都很熟。 植物单细胞的阶段要开启了,今天提到的这篇文章是国内首篇,也是世界范围内的第四篇植物单细胞文章。除此之外,还有几篇已经在 bioRxiv 上上线了,不过还没有正式发表。这些文章都不约而同选择了植物中研究最广的模式之物拟南芥,而且全部研究的是根尖。后面在发展就要看看其它组织和物种的情况了。 如果一个东西在植物里已经出现要快速增长的趋势,那么它在人和动物里应该也就已经相对比较成熟了。此时,即便暂时还用不到,但是相关的技术和方法就需要留意和学习起来。 在这篇文章中,作者在单细胞水平揭示了拟南芥根尖细胞的异质性重构了根尖分生组织细胞的发育轨迹。按照文章的说法,成功拿到了 7695 个根单细胞转录组数据。聚类后根细胞被划分为 24 个细胞类群,细胞类群注释分析鉴定了一些潜在的新细胞类型,并找到了一批细胞类型标记基因。 用 t-SNE 和 UMAP 重构了根发育的基本轨迹,实现了根分生组织细胞分裂和分化在单细胞水平上的准确投影。进一步利用伪时间(pseudo-time)分析,捕获了根尖分生组织细胞的分化轨迹和过渡态细胞,解析了根分生组织细胞如何通过协调细胞分裂和分化进程逐步形成根尖不同细胞类型的分子机理。此外,通过分析细胞类群对离子吸收和激素响应情况,揭示了不同根细胞类群的响应热图。该研究加深了我们对拟南芥根细胞组成和发育轨迹的认识,将根发育生物学从原先的组织器官水平提升到了单细胞水平。 (上面这一段话主要来自官方报道,具体的细节需要仔细读完文章在分享) 目前已经正式发表的其它三篇植物单细胞文章,看下来会发现分析思路和 figure 都异曲同工。 DOI(url): https://doi.org/10.1101/611137 发表日期:April 16, 2019. 有哪些 alignment-free (AF) 相关的工具,以及如何评价。 AF 类的工具,在转录组分析层面使用最多的是定量分析。例如 salmon 和 kallisto,主要原理就是基于对 kmer 的各种操作。其实除了转录组的快速定量 这篇文章比较详细的介绍了目前主要 AF 相关工具的原理和工具。同时,作者使用了 24 个相关软件的 74 种方法,测试了五种应用场景,分别是: 作者还提供了一个在线工具,用来展示这些结果。 DOI(url): https://doi.org/10.1186/s13059-019-1688-1 发表日期:18 April 2019 bam 文件似乎可以方便的回滚了 随着参考基因组的更新和比对方法的更新,很多之前的 bam 文件似乎就变得过时了。除了找出原始的 fastq 文件再重新来过一次,现在有了另一个选择。 整体而言,和目前已有的一些可以转换 bam 文件的工具相比,其在内存和存储占用,已经方便程度上都有优势。 目前我的问题是很多时候会对原始 bam 文件进行一波过滤,这个时候已经丢掉了很多 fastq 的 reads。 其它几个已有工具的比较 DOI(url): https://doi.org/10.1105/tpc.18.00601 发表日期:March 2019 在我看来,假基因和 lncRNA 这类非编码 RNA 其实大多数是定义方式不同,重合度不低。 假基因(Ψs)一般是和功能基因的序列相近的非功能性基因,通过复制或逆转录方式形成,通常会含有各种突变导致基因功能的丧失。在这篇文章中,作者检查了七种被子植物(拟南芥,短柄草,大豆,苜蓿,水稻,杨树和高粱)假基因的起源,进化和表达模式及其与非编码序列的关系。作者鉴定了大约 250,000 个假基因,发现非常大比例的非转座因子调控非编码 RNA(microRNA 和 lncRNA)起源于假基因近端上游区域的转录。 还发现与随机基因间区相比转录因子结合位点优先发生在假基因近端上游区域,这表明假基因可能通过提供用作启动子和增强子的转录因子结合位点来调节基因组进化。 假基因定义流程: 假基因鉴定情况 主要鉴定步骤 以及 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名 - 非商业性使用 - 禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。A Single-Cell RNA Sequencing Profiles the Developmental Landscape of Arabidopsis Root
关键点
参考意义
相关内容
Benchmarking of alignment-free sequence comparison methods
关键点
参考意义
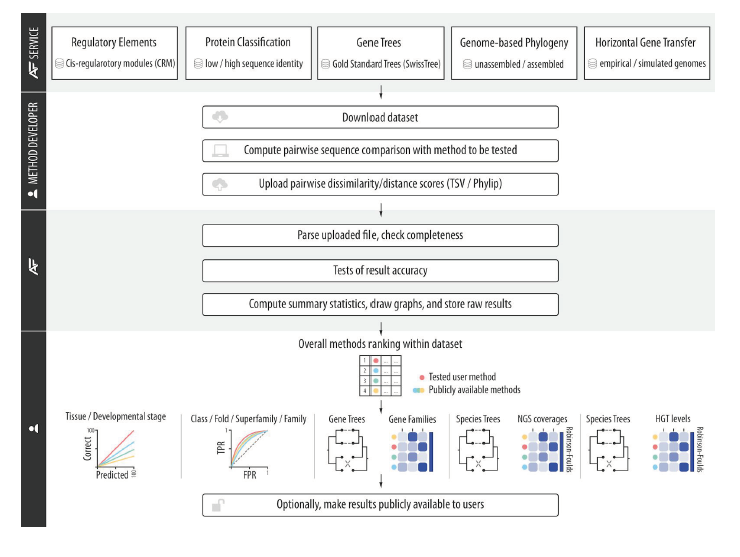
相关内容
Bazam: a rapid method for read extraction and realignment of high-throughput sequencing data
关键点
参考意义
bazam 首先可以从 bam 或者 cram 文件直接找到 pair reads 在比对回其它参考基因组而不需要中间步骤; 也可以按需要提取过滤后的 reads,例如与特定基因位置有 overlap 的 reads。结果可以直接传到下游工具,或以 fastq 格式存储以供进一步处理。另外还从 read 这个 input 层面提供了多线程比对的思路。加快了比对速度。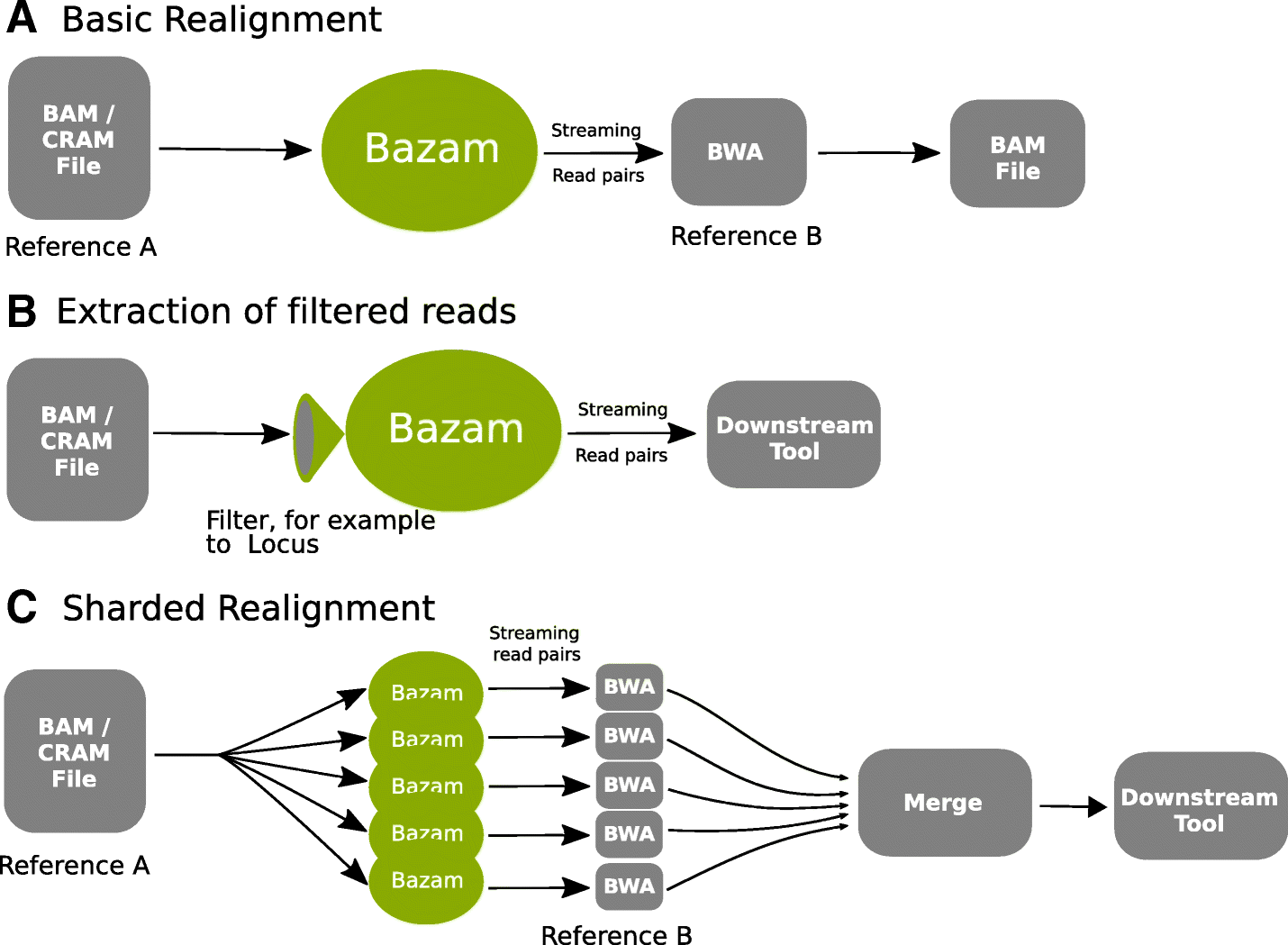
相关内容
Tool
Storage used
Memory
Effective Cores
Time
Sort-Extract-Realign
282 GB
20 GB
16
13 h, 15 min
Picard SamToFastq
148 GB
78 GB
16
16 h, 14 min
Biobambam bamtofastq
149 GB
30 GB
16
15 h 30 min
Bazam (no sharding)
68 GB
28 GB
16
14 h, 55 min
Bazam 10-way sharding
102 GB
20 GB
160
1 h, 11 min
Evolutionary Origins of Pseudogenes and Their Association with Regulatory Sequences in Plants
关键点
参考意义
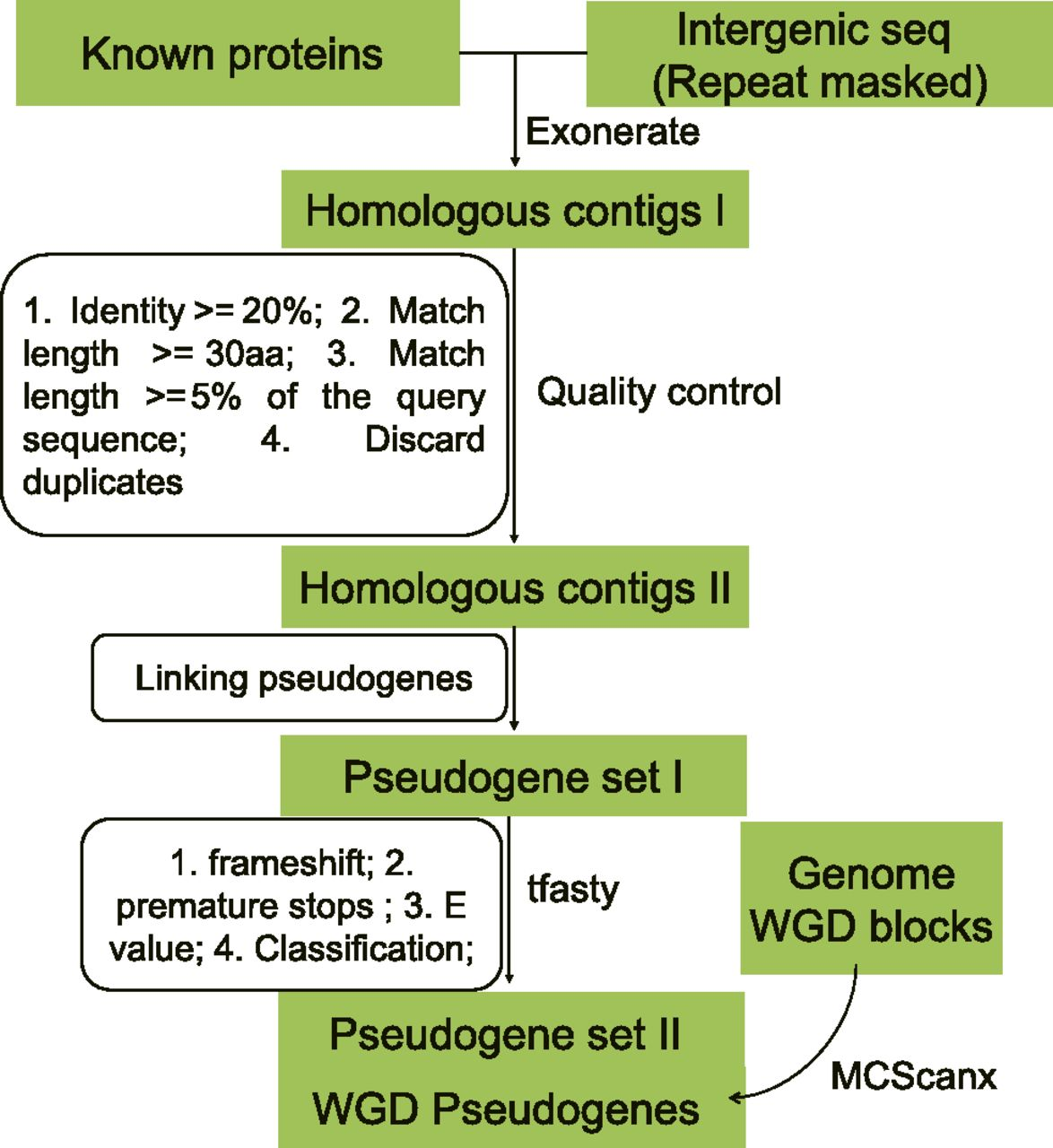
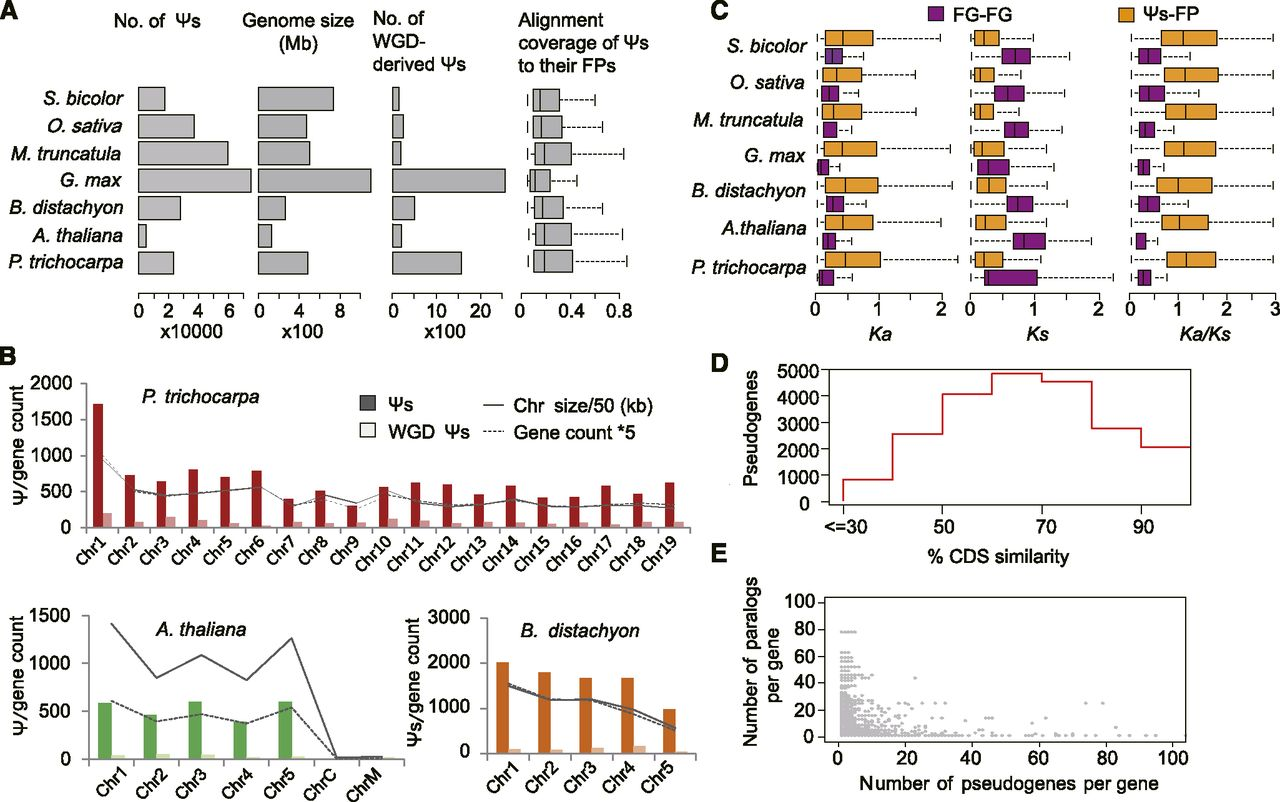
相关内容
--model protein2genome --showquerygff no --showtargetgff yes --maxintron 5000 --showvulgar yes --ryo \"%ti\\t%qi\\t%tS\\t%qS\\t%tl\\t%ql\\t%tab\\t%tae\\t%tal\\t%qab\\t%qae\\t%qal\\t%pi\\n\".
-A -m 3 q
-k 50 -g -1 -s 5 -m 25
· 分享链接 https://kaopubear.top/blog/2019-04-19-weeklypaper/