DOI(url): https://doi.org/10.12688/f1000research.17916.1 发表日期:24 Feb 2019 差异表达中比较少见的一种方式 在 F1000Research 发表的这篇文章介绍了一个利用 alignment-free RNA-seq quantifications 结果进行差异分析的工具,主要是用来对转录本进行差异定量分析。在日常的分析中我们进行差异分析最常见的是对差异基因进行定量分析,这里没有考虑到每个基因内部转录本的情况。 在这篇文章中提到了一个三种差异分析的方法,分别是: 根据需求的不同,这三种方法会分析出非常不同的结果。在 DTU 中,即便是两个表达总量没有差异的基因其也可能发生 isoform switching,及 dominant isoform 的改变。而文章中作者发表的 R 包即可进行 DTU 的分析,其输入数据可以是 Kallisto 或者 Salmon 的定量结果。 R 包地址:https://github.com/bartongroup/Rats 考察一个基因内不同转录本在不同情况下的表达丰度,这个需求在我的实际分析中还没有用到。不过把转录本定量的结果转为基因定量的结果,使用 R 包 tximport。 DOI(url): https://doi.org/10.1101/585745 发表日期:March 22, 2019 详细讨论转录组 de novo 拼接的那点事。 这篇发表在 biorxiv 的文章,从多个方面阐述了转录本拼接本身存在的问题。这其中包括作者评估的几个算法都没有拼出数百个真是表达的基因,一大部分拼接处的 contigs 完全由内含子和 UTR 组成;对转录本有效长度的不准确给定量带来了很大的偏差等等。最后建议现在测序价格便宜了,能拼基因组就拼基因组吧。(这道理难道我不懂么,我只是没钱)。 这篇文章更值得参考的是他做的分析和采用的方法,50 页的文本非常详细的记录了具体的分析过程以及参数。也提醒了我们在做类似的分析是有哪些角度可以思考。 主要分析流程 敢把流程写的及其详细的文章自然就敢把代码全部 show 给你,GitHub 地址 TranscriptomeAssemblyEvaluation 。 DOI(url): https://doi.org/10.1101/581306 发表日期:March 18, 2019 非编码区 SNP 可以研究的方向之一,对转录因子结合能力的影响。 GWAS 显示 88%的疾病相关 SNP 位于非编码区。然而,非编码 SNP 仍未得到充分研究,这其中一部分原因是它们难以确定实验验证的优先级。这篇文章作者提出一种确定非编码区 SNP 重要性的评判标准以及方法。通过观察全基因组功能性转录因子结合位点内 SNP 的 ChIP-seq 信号强度差异来估计转录因子结合亲和力 目前用来分析转录因子结合位点的 martix 叫做 position weight matrix (PWM),而文章中则提出了 SNP effect matrices (SEM)。 SNP Effect Matrix pipeline 使用数据 流程 GitHub https://github.com/Boyle-Lab/SEM_CPP DOI(url): https://doi.org/10.1101/580829 发表日期:March 19, 2019 针对 Bulk segregant analysis (BSA) 设计的无参考基因组 call snp 流程 对于植物(作物)来说,通常手里拿到的材料并不是真正的测序品种。比如水稻就包括籼稻和粳稻,而且很可能手里的粳稻也不是真正的粳稻。面对这种情况,其实不少用到和参考基因组比对的工作都或多或少的存在一些问题,因为那个基因组在很多位置其实你都无法「参考」,而其中影响最大的一类工作就是 call snp 相关的工作。当你的材料和参考基因组差别比较大的时候就会存在很多黑箱。如果能够不依赖基因组信息而直接 call snp 一定程度上就可以规避这类问题。但是准确度如何需要进一步考证。 相关的几篇文章: 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名 - 非商业性使用 - 禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。文献题目 Relative Abundance of Transcripts (RATs): Identifying differential isoform abundance from RNA-seq
关键点
参考意义
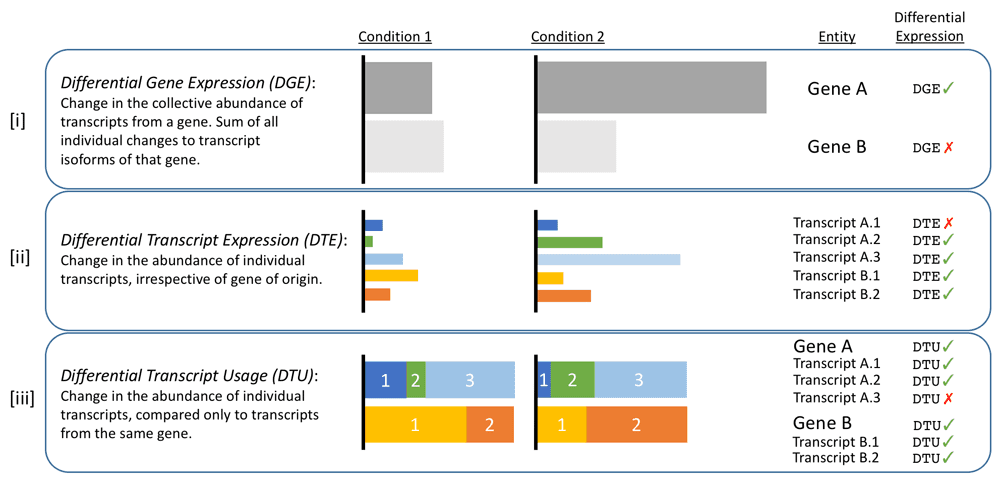
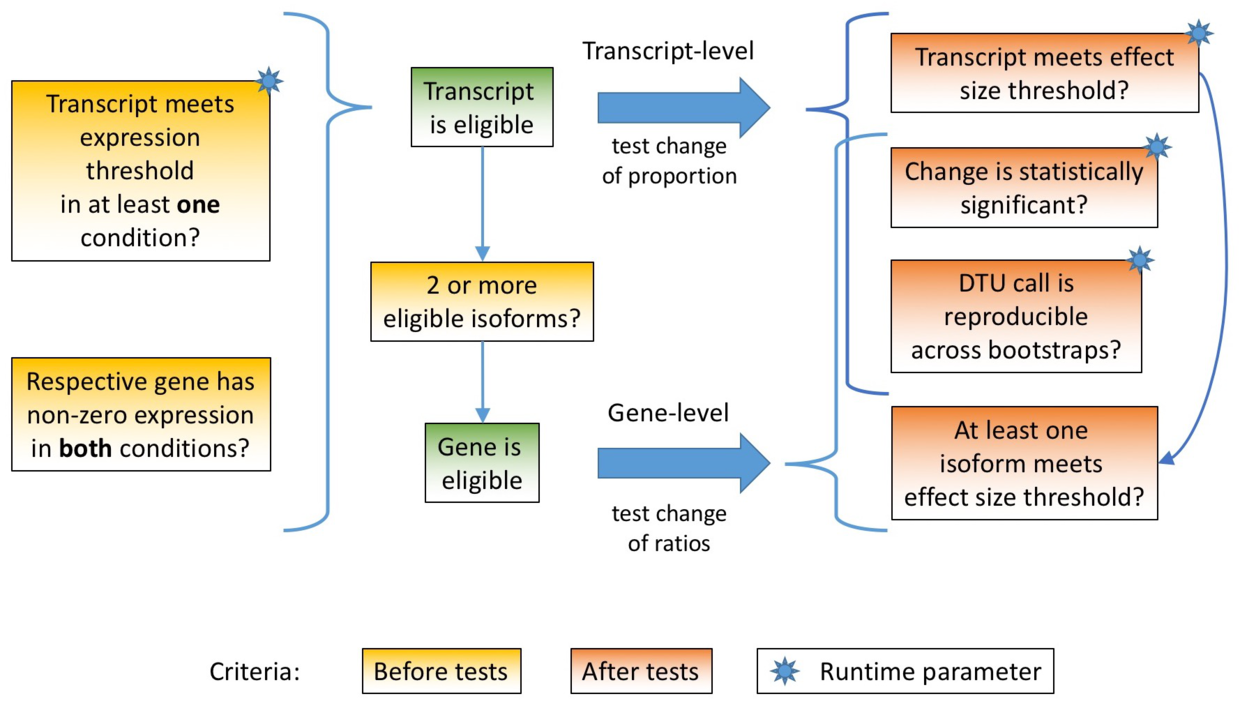
相关内容
文献题目 Error, noise and bias in de novo transcriptome assemblies
关键点
参考意义
相关内容
文献题目 Predicting the effects of SNPs on transcription factor binding affinity
关键点
参考意义
的变化。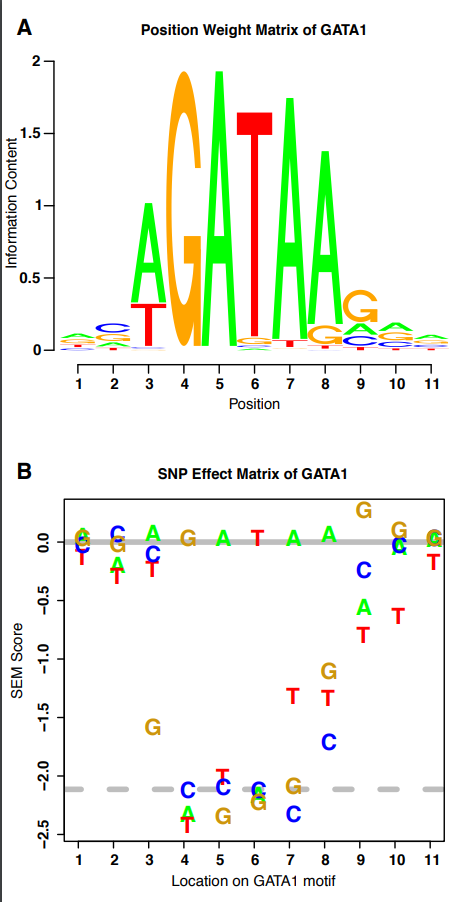
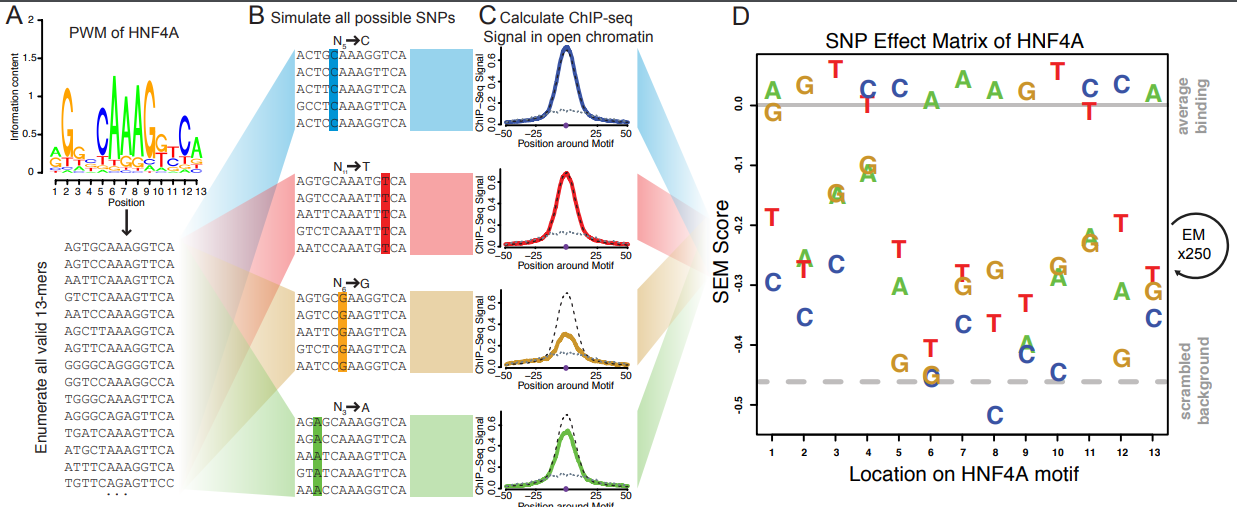
相关内容
文献题目 LNISKS: Reference-free mutation identification for large and complex crop genomes
关键点
参考意义
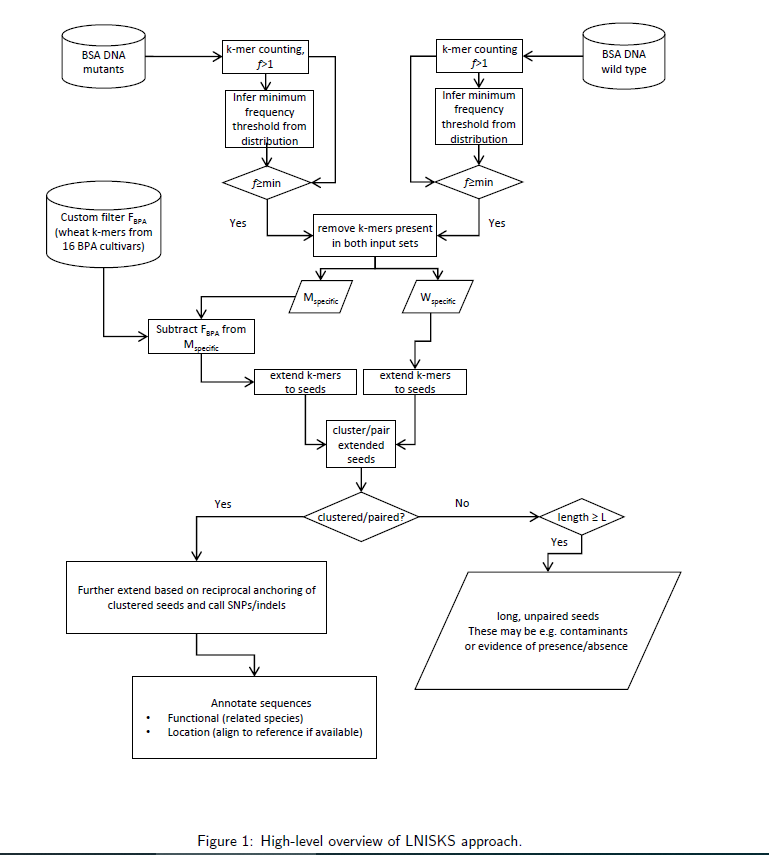
相关内容
· 分享链接 https://kaopubear.top/blog/2019-03-30-weeklypaper/