如果让你说出日常在进行生物数据分析时,做的最多的事情是什么,我想不管你是什么方向,「不停地转换格式」应该能排进日常前三。如果再问用的最多的工具是什么,想必一定也是和「不停地转换格式」相关的工具。 在这些工具里有几个叫 XXtools 的非常出名,其中 samtools 和 bcftools 我们这次暂且不提,它们都是出自大神 LiHeng 之手。另一个能脱口而出的恐怕就是 bedtools 了,这个工具最初发表在 Bioinformatics 的文章,谷歌学术显示自 2010 年以来已经有了 6600 多次引用。当年这篇文章的第一作者 Quinlan ,如今已经是 UNIVERSITY OF UTAH 很厉害的 PI,它的课题组先后开发了一些列优秀的生物信息工具。当然,他们的故事今天也暂且不提(那位朋友问了,你今天到底要说啥,且看下文)。 6600 多次引用(其实现在很多文章用了它已经不引用了)让 bedtools 在 bed 相关格式文本处理领域一家独大。我经常半开玩笑地说 50% 高通量数据后期分析概括一下都是各种位置之间的纠缠,因为但凡分析有参考基因组的数据就会把所有信息都铆定到参考基因组这个坐标系中(如果没有基因组那就拼一个),这也是为什么 bedtools 从一推出就如此受欢迎。在生物信息这个领域,一个软件是不是会被大量使用,最重要的是它是否真的有用然后再加上一点点出现的时机和运气。 除了 bedtools,其实还有一个处理 bed 系列文件很强大的工具没有引起太多人的注意,这个软件叫做** bedops**。比 bedtools 晚出生两年,同样发表在 Bioinformatics,至今它的文章只有 273 次引用。当然这里的「只有」是和 bedtools 进行对比而言,不要误会。 bedops 突出的特点是多快好省。它的运算速度要比 bedtools 快不少(后来 bedtools 升级速度有所改善)同时针对大文件很省内存,而且支持各种 pipe 的结合连用。具体可以了解 官方文档,其中作者专门做了一个「你问什么应该使用 bedops」的解释。链接如下: 这篇文章不会对 bedops 的功能逐一介绍,但是从今天开始你的心里要记住一件事:bedtools 中 95% 的功能在 bedops 中都有对应的方法(有些实现的更好),而且 bedops 还有一些其他特有且好用的命令。另外 bedops 有一个的隐形有点也需要记住,如果有一天你在使用 bedtools 处理某些大基因组数据,因为超过软件本身的各种限制而报错无法使用时,用 bedops 吧,它不存在这类问题。 总之,从今以后,你的心里不该只有 bedtools。 考虑到文章篇幅,接下来只为大家介绍一种日常分析比较常见的需求供参考:对两组 bed 相关文件进行统计计算。 具体分析场景: 已有两组感兴趣的位置文件,至于这些位置是从哪来的都无所谓,可能是某组基因所代表的坐标,可能是某些分析找出的结合位点,还可能是一些 snp 或者 indel 的变异信息。有了这两组位置信息,如果只是想知道一些交并补集的逻辑运算在 bedtool 中常用的 intersect, closest, merge 等等就可以完成。但是,如果想根据一组位置,对另一组位置对应的某些数据进行相对复杂些的统计计算要如何操作呢? 比如想知道在 1024 个上调基因位置区间(一组位置)内每个基因里有多少个某某转录因子的 peak,每个区间这些 peak 的最大值是多少,亦或者这些 peak 峰度均值和变异系数(另一组位置对应的某些数据)是什么。 在 bedtools 中有一个对应的命令叫做 groupby, 官网文档对这个命令的基本解释如下: Given a file or stream that is sorted by the appropriate “grouping columns” (-g), groupby will compute summary statistics on another column (-c) in the file or stream. This will work with output from all BEDTools as well as any other tab-delimited file or stream. 目前, 具体的使用示例去参考官方文档吧,只提一句,一般 groupby 和 intersect 连用较常见。 针对上文提到的需求,在 bedops 中有一个对应的核心工具叫做 一个典型的 map bed 文件格式如下: 第一类是处理参数,例如指定处理速度(是否开启快速处理)和数据格式等;第二类是 overlap 参数,即满足怎样的条件才进行计算,例如指定位置间 overlap 的碱基数或者比例等,这一部分就比 bedtools 灵活很多;第三类参数则为运算参数,其中一个子类是分值类计算,例如最大值,均值,中位数和变异系数等等,另一个子类是非分值类参数,例如包含个数等。需要提醒,如果进行分值类运算 map bed 需要 5 列,如果是非分值类运算 map bed 文件根据要计算的内容不同还略有差别。更多的使用方法和技巧有机会就再写写,如果你感兴趣还是去学习官方文档。 就个人使用体验来说,我目前已经把绝大部分 bed 文件相关处理工作从 bedtools 转移到了 bedops,现在也把这个工具推荐给你。 当然,除了上面两个工具,在 Python 和 R 中也有人进行了相应的移植工作以满足不同的使用环境和需求,在文章的最后列出来供你参考。 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。bedtools 的傲人光环
bedops 略被忽视的优秀工具
https://bedops.readthedocs.io/en/latest/content/overview.html#why-you-should-use-bedops试举一例供你参考
使用 bedtools groupby
groupby 主要参数为 -g、-c 和 -o。其中,-g 指定根据那些信息进行 group,默认是根据 bed 文件的 1-3 列; -c 指定针对哪一列数据以 group 为单位进行各种运算; -o 则指定进行何种运算。groupby 可以进行的运算内容如下:sum - numeric only
count - numeric or text
count_distinct - numeric or text
min - numeric only
max - numeric only
mean - numeric only
median - numeric only
mode - numeric or text
antimode - numeric or text
stdev - numeric only
sstdev - numeric only
collapse (i.e., print a comma separated list) - numeric or text
distinct (i.e., print a comma separated list) - numeric or text
concat (i.e., print a comma separated list) - numeric or text
freqasc - print a comma separated list of values observed and the number of times they were observed.
Reported in ascending order of frequency*
freqdesc - print a comma separated list of values observed and the number of times they were observed.
Reported in descending order of frequency*
first - numeric or text
last - numeric or text
使用 bedops bedmap
bedmap,它可以根据你的基因组输入文件提供各种各样的统计操作。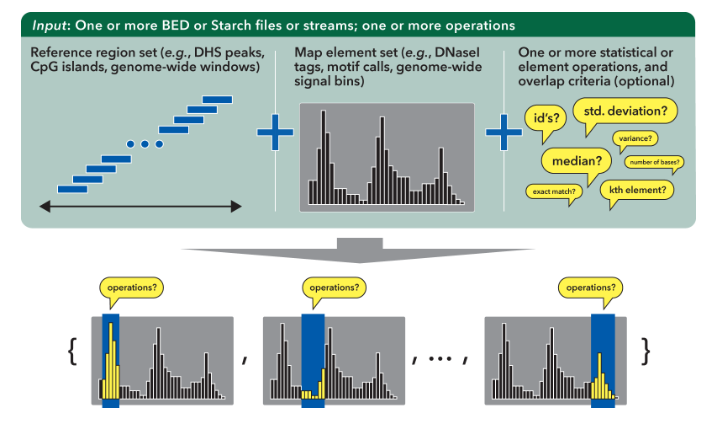
bedmap 的输入文件为两个 bed 文件,其中一个用来分 group 的叫做 ref-bed,这个文件可以是最简单的三列格式,用来提供位置信息;另一个需要进行后续计算的 bed 文件叫做 map-bed ,它根据实际计算需求要提供 4 列或者 5 列的 bed 文件,其中 1-3 列为位置信息,第 4 列为 id,第 5 列为要计算统计的 value。chr21 33031165 33031185 map-1 1.000000
chr21 33031185 33031205 map-2 3.000000
chr21 33031205 33031225 map-3 3.000000
chr21 33031225 33031245 map-4 3.000000
chr21 33031245 33031265 map-5 3.000000
chr21 33031265 33031285 map-6 5.000000
chr21 33031285 33031305 map-7 7.000000
bedmap 参数整体可以分为三大类,如下所示:USAGE: bedmap [process-flags] [overlap-option] <operation(s)...> <ref-file> [map-file]
写在最后
· 分享链接 https://kaopubear.top/blog/2019-03-03-bedopsandbedtools/