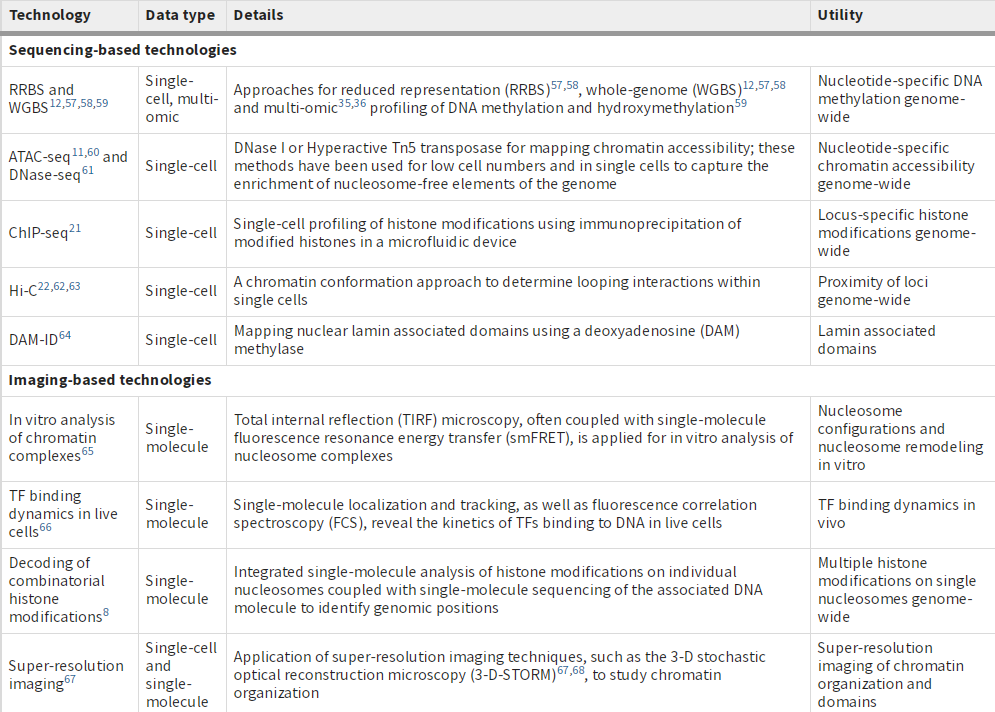
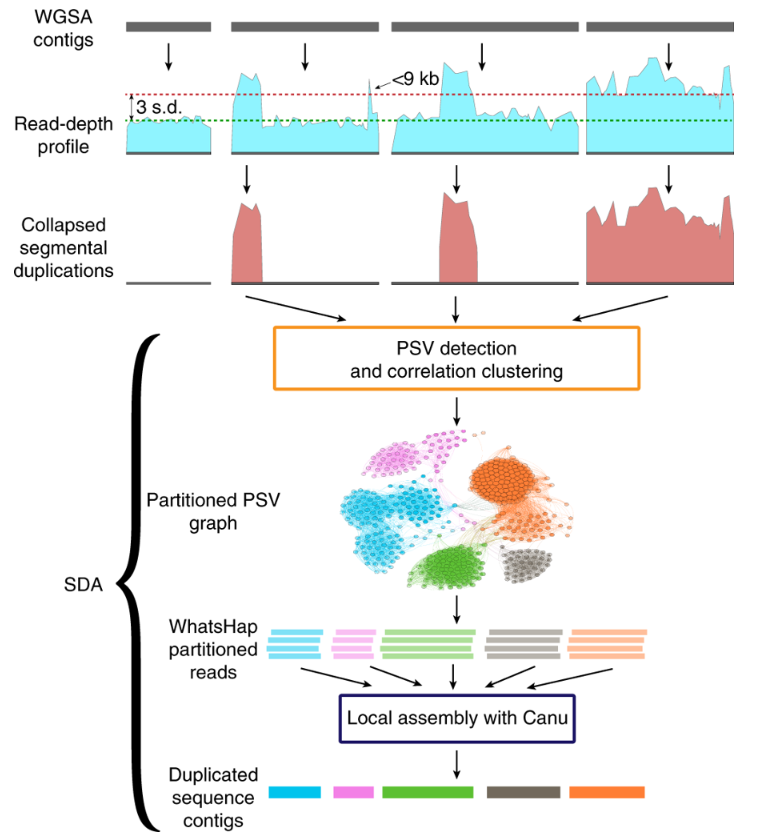
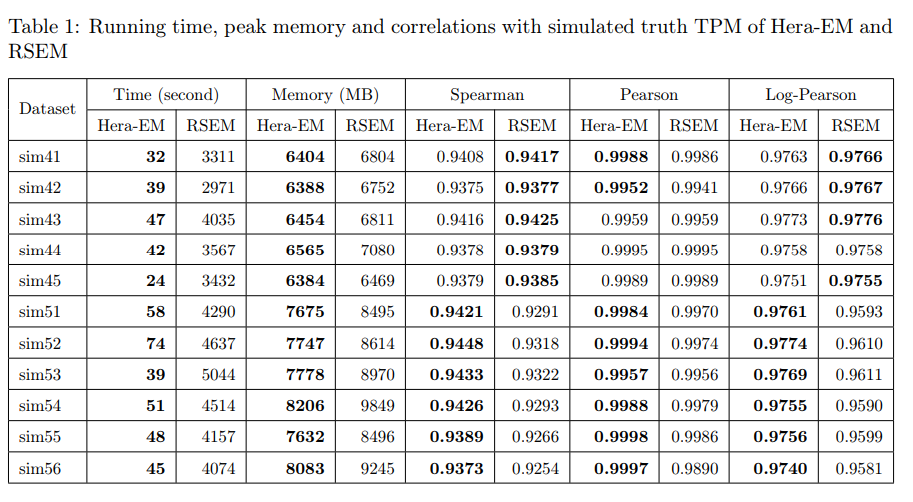
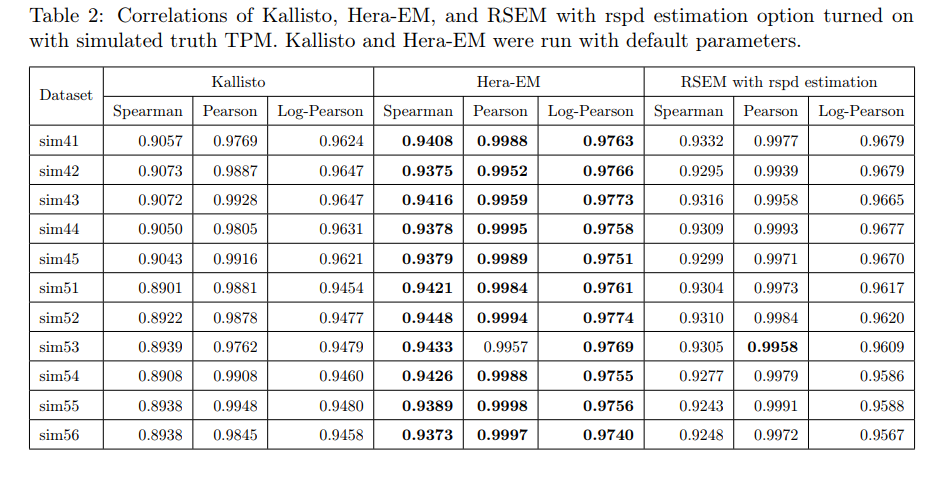
文献题目:Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution DOI(url): https://doi.org/10.1038/s41588-018-0290-x 发表日期: 2018 Dec 17 这篇综述介绍了单细胞和单分子表观基因组学正在以前所未有的分辨率揭示基因组调控的各种机制。作者重点介绍了变革技术中的一部分,并讨论了它们如何被用于识别新的基因调控模式 这些技术如下表所示 作者将这些分析与单细胞转录组学的最新进展进行了对比,论证了表观基因组学技术在理解细胞多样性和发现基因调控机制中的重要作用。 另外这边文章的「Box」作者还首先阐述了进行表观研究需要测定的一些信息。如果你对表观遗传完全没有概念推荐了解。 文献题目:Long-read sequence and assembly of segmental duplications DOI(url): https://doi.org/10.1038/s41592-018-0236-3 发表日期: 2019 Jan;16 作者开发了一种基于长序列 reads 的多倍体 Pasing 计算方法,用来解决基因组组装中大片段重复的折叠区域问题。 方法流程图: 该方法利用旁系同源序列突变(PSV)和相关性聚类来精确组装先前在 long-read 人类基因组组装中塌陷的的 SD 的不同旁系同源大片段重复序列。 首先利用全基因组重测序数据根据深度的差异定义大片段的重复区域,其中大于 9kb 且覆盖度高于 3 个 sd 加上均值并且不全部是由 common repeat 组成的区域被认定为 SD ;接下来定义每个基因组塌陷区域的旁系同源序列突变 paralogous sequence variants (PSVs) 并对 PSV 进行聚类。随后根据 PSV 利用 WhatsHap 将 reads 进行分组然后分别对不同组的 reads 进行组装。 在基因组异染色质和基因富集区域存在很多大片段重复 segmental duplications (SDs),长度大于 100kb 并且存在于多个位置。即便是基于 FALCON 的单分子实时(SMRT)测序的基因组组装在很多结果中也有一半类似的区域没有解决。 当前 ONT (https://nanoporetech.com/) 和 PacBio (https://www.pacb.com/) 的主要问题是测序的错误率会达到 10%-15%,主要针对 WGSA 的长序列组装方法是基于 reads 的矫正和矫正后的重叠来得到更长的 contig。但是这些高错误率对于区分旁系同源和等位基因序列特别成问题,因为很多序列重复序列高度相似(> 95%)并且差异部分在测序的误差范围内。这篇发表在 nature methods 文章提供了一个不错的思路。 工具地址:Segmental Duplication Assembler (SDA; https://github.com/mvollger/SDA) 文献题目:A revisit of RSEM generative model and its EM algorithm for quantifying transcript abundances. DOI(url): https://doi.org/10.1101/503672 发表日期: December 21, 2018 (预印本) 本文作者团队修改了 RSEM 的 EM 算法,而且将计算时间减少了大约 100 倍,而且准确性还略有提高。 以下是一些比对测试信息 RSEM 定量相对其他软件是比较准确的但是速度比较感人,近几年 Salmon, Kallisto 这类软件因为采用了 pseudoalignment 极大的提高了运算速度也取得了很大的成功。但是,这类软件不能提供一些具体的比对信息,导致很多需要这些比对信息的软件无法进行。 现在,有团队出来管管这事儿了,不过实际的使用感受和测试还没有进行。另外,他们作为一个商业团队还有一个开源的 RNA-seq 分析,具体信息可以参考相关内容里的链接。 hear https://github.com/bioturing/hera hear-EM https://github.com/bioturing/hera/tree/master/hera-EM 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。单细胞及单分子在表观的应用
关键点
参考意义
相关内容

一种组装大片段重复区域的方法
关键点
参考意义
相关内容
RSEM 转录本定量的提速升级版
关键点
参考意义
相关内容
· 分享链接 https://kaopubear.top/blog/2018-12-23-weeklypaper/