针对转录组数据,平时分析中最常见两组之间的比较,比如不同处理或者不同突变体。面对这样的数据用用 DESeq2 或者 edgeR 基本就差不多。如果样本量或者不同条件很多的话可以还做 WGCNA 的分析。但是生物体的生长发育和时间这个维度有着非常密切的关系。如果碰上了一组和时间有关系的数据可以怎么处理呢? 所谓时序分析 (time series analysis) 在 data science 中是非常重要的一个方向。对大多数商业行为而言如果能够通过已有不同时间数据来进行预测就有可能大大提高自己的胜率。通常时间序列数据会包括趋势部分和不规则部分, 我们需要做的就是剔除不规则部分然后找到趋势所在,再进行预测。在预测过程中通常可以采用移动平均法、局部加权回归法、指数平滑法和自回归整合移动平均等方法。 生物学的时间相关数据本身预测属性和商业数据相比要弱很多。一种是单一条件的纯时间序列,主要看不同基因的表达模式,根据相似的表达谱将基因归为多个类有助于找到功能相似的基因。另一种情况是含有对照和处理的时间序列,需要再考察不同条件的差异基因。 关于时间序列转录组数据分析的工具,近三年来有两篇偏综述和测评类的文章(一个人写的)。 在这两篇文章中还是提到了一些工具,但其中有一些用到 matlab(这软件贵啊),有一些年久失修或者不维护或者和最新 R 版本不兼容,筛筛捡捡能用的且文章里认为还不错的也就剩下三四个。 来自于 DESeq 的方法,下文中提到的 ImpluseDE2 和 MaSigPro 都使用了这种模型。 来自于 maSigPro 方法,所谓多项式回归区别常见的线性回归,会把一次特征转换成高次特征的线性组合多项式,比使用直线拟合更加准确。但是到底用几次方需要具体分析,次数过高会出现过拟合。在能够解释自变量和因变量关系的前提下,次数应该是越低越好,这也算是奥卡姆剃刀原则吧。 所谓自回归是统计上一种处理时间序列的方法,用 至 来预测本期 的表现并假设它们为线性关系。简单说就是用自己来预测自己,因为是从回归分析中线性回归发展而来只是用 x 预测 x,所以叫自回归。 同样是来自于 DESeq 的方法,下文中提到的 ImpluseDE2 和 MaSigPro 也都使用了这种方法。 似然比检验 (likelihood ratio test,LRT) 用于比较两个模型的拟合优度进而确定哪个模型与样本数据拟合的更好。其中一个是具有一定数量项的完整模型,另一个是删掉完整模型中一部分项的简化模型。LRT 检验中,自由度等于在简化模型中减少的模型参数数目,LR 近似符合卡方分布。一个相对复杂的模型与一个简单模型比较,如果可以显著地适合一个特定数据集,那么这个复杂模型的附加参数就能够用在以后的数据分析中。 The LRT examines two models for the counts, a full model with a certain number of terms and a reduced model, in which some of the terms of the full model are removed. The test determines if the increased likelihood of the data using the extra terms in the full model is more than expected if those extra terms are truly zero. 为了测试多个时间点的任何差异,可以使用包含时间因子的设计和时间因子在简化公式中被删除的另一个设计。对于包括对照和实验组的时间序列,可以使用包含条件因子,时间因子和两者相互作用的公式。在这种情况下,使用具有不包含相互作用项的简化模型的似然比检验将测试该条件是否在参考水平时间点(time 0)之后的任何时间点可以诱导基因表达的变化。 EBseq-HMM 采用的方法,来自于 BEseq。 这个软件最早发表在 2007 年,相对老一些好在目前仍然在维护,其主要目的是给时序数据进行基于模糊聚类算法的聚类。我们常见的聚类算法可以分为严格聚类 (hard clustering) 和模糊聚类 (Fuzzy clustering )(也叫做宽松聚类 soft clustering)。严格聚类会将一个基因只聚到一类中,kmeans 就属于严格聚类。而模糊聚类允许同一数据属于多个不同的类,其聚类结果是一个数据对聚类中心的隶属度,0 到 1 之间。对于分类很开的数据使用严格聚类是没问题的。但对于时序表达量数据来说,不同的类常常会有重叠,所以可以尝试宽松聚类方法。算法需要首先设定一些参数,若初始化参数不合适,可能影响聚类结果的正确性。 在使用 Mfuzz 时首先应该进行数据标准化处理 ,可以使用类似于 FPKM 或者 TPM 的表达结果也可以使用 DESeq2 矫正后的结果进行比较分析,另外不支持值为 0 的数据,所以需要加上 pseudocount 。除此之外,Mfuzz 接受的数据格式为 ExpressionSet,需要对矩阵进行转换。 这个包只能进行聚类,是找不了有处理对照组的差异基因的。需要注意。 关于上面的 R 代码,我深知在 R 中写 for 循环的丑陋,但是目前还没有掌握向量结构化处理变量和输出的方法。 运行 masigpro 主要有四步: 有两点内容需要注意:对于无对照的单一时序数据处理方法;以及处理转录数据时的特殊参数。因为这个包不会对数据进行标准化,所以应该提前做好,使用 DESeq2 即可。 另外,在实际分析的时候可能会出现 ImpulseDE2 是最近才出来的一个 R 包,在前面提到的综述评测文章中认为这个包找时序数据中的差异基因效果最好,它可以用来解决两类问题。 这个包中,有一个 在展示的热图中会出现四部分,包括 transient and transition trajectorie,其中每一种 tarjectorie 又包括 up 和 down 两类。所谓的 transient 可以理解为时序数据在中间某一个时间点存在 up 或者 down peak,即在某一个时间点存在表达的最大或者最小值;而所谓的 transient 可以理解为一个持续的变化,比如持续的升高或者持续的降低。 EBSeq-HMM 是基于 EBSeq 二次开发的工具,主要用于分析时序数据。在计算的时候首先基于负二项分布对参数进行估计,然后利用自回归隐马模型将基因的表达进行分类。比较神奇的是,最终给到的结果会标示为 因为目前做的数据是没有对照的单一时间序列数据,所以还不能体会哪一个找出的差异基因更准确些。但是如果只是想把所有的基因根据不同的时间点分为若干表达 pattern,似乎结合 Mfuzz 和 ImpulseDE2 就可以了。 当然,涉及到聚类,尤其是非监督聚类的时候通常主观因素还是较强,如果能对关键基因或者数据有一个大致的估计预判操作起来会相对轻松些,如果没有,可能就需要结合不同类的生物学意义等角度来找合适的聚类数目了。 http://a-little-book-of-r-for-time-series.readthedocs.io/en/latest/src/timeseries.html 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。
时序分析
生物学时序分析
可用的分析时间序列工具
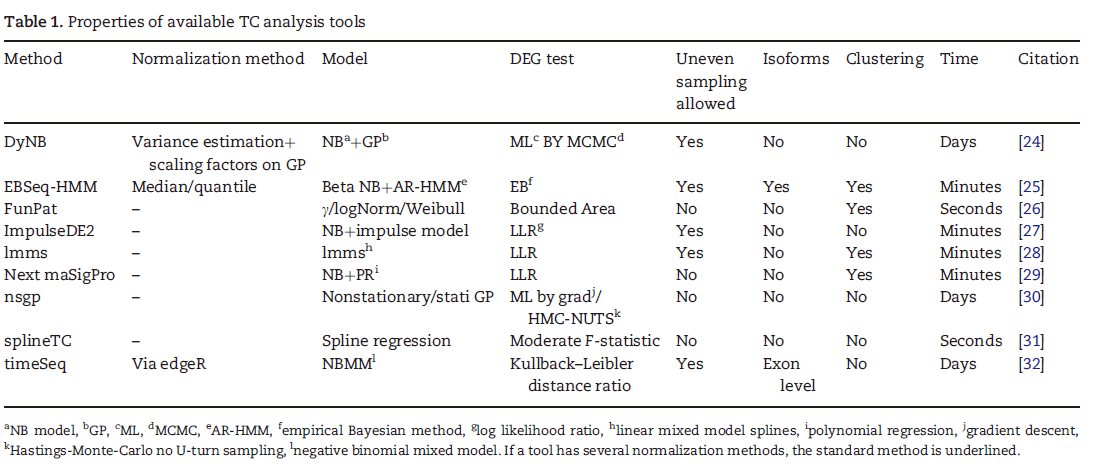
主要模型
差异基因检测方法
具体应用
Mfuzz
library(Mfuzz)
library(RColorBrewer)
# 读取数据
tpm <- read.table("sample_tpm_mean.txt", header=T, stringsAsFactors=F)
# 过滤极端值
tpm <- tpm[rowMeans(tpm)>1,]
tpm <- tpm[rowMeans(tpm)<10000,]
# 添加 pseudo tpm
pseduo <- 0.01
tpm <- tpm+pseduo
tpm <- as.matrix(tpm)
# tarns2Eset
tpminput <- new('ExpressionSet', exprs=tpm)
# 过滤变化小的数据
tpminput <- filter.std(tpminput, min.std=1)
# 类似于 z-score
tpminput.s <- standardise(tpminput)
# 分不同个数的进行分析,提取高可信度基因并画图然后输出每个类的结果
for (i in seq(8,16,2)) {
assign(paste("tpmcl", i ,sep = "_"),
mfuzz(tpminput.s,c = i, m = mestimate(tpminput.s)))
assign(paste("tpmclfilter", i ,sep = "_"),
acore(tpminput.s, cl = get(paste("tpmcl", i ,sep = "_")), min.acore = 0.5))
pdf(file = paste("mfuzz",i,"cluster.pdf",sep = "_"), width = 12, height = 8)
mfuzz.plot2(tpminput.s,cl = get(paste("tpmcl", i ,sep = "_")), mfrow = c(3,3),
centre = T,time.labels = c(paste(c(0,1,2,4,8,12),"h")),
colo = brewer.pal(9,"Blues"), x11 = F)
dev.off()
for (k in 1:length(get(paste("tpmclfilter", i ,sep = "_")))) {
write.table((get(paste("tpmclfilter", i , sep = "_")))[[k]],
paste("mfuzz", i ,"cluster", k,"gene.txt", sep = "_"),
quote = F, sep = "\t", row.names = F, col.names = T)
}
}
MaSigPro
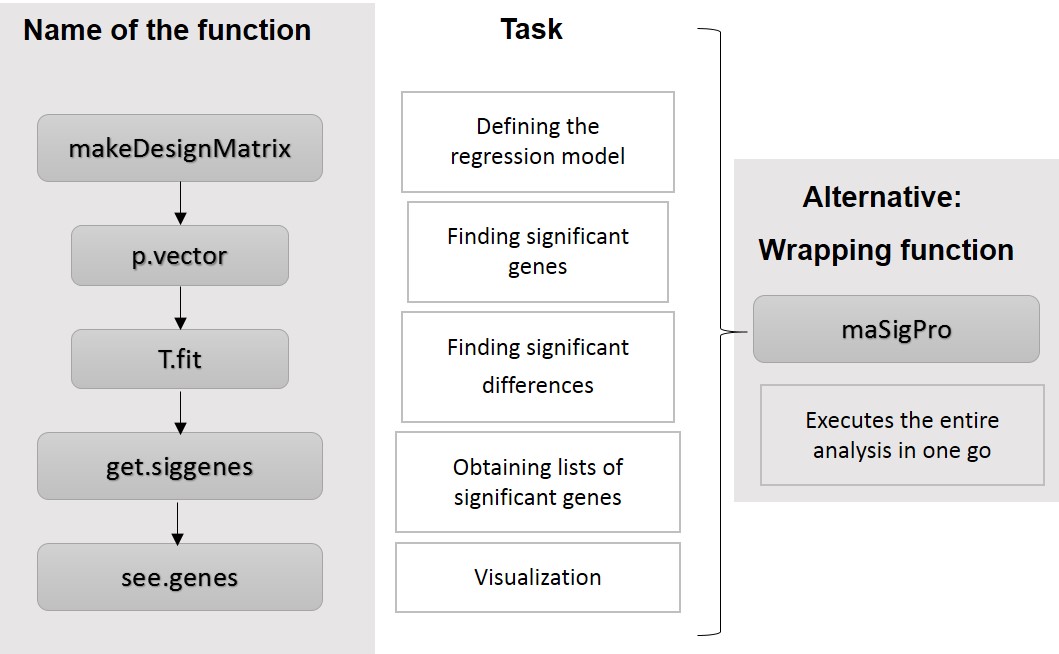
glm.fit: algorithm did not converge 的警告。这是由于进行 logistic 回归时,依照极大似然估计原则进行迭代求解回归系数,glm 函数默认最大迭代次数是 25,当数据不太好时 25 次迭代可能还不收敛,一方面可以增大迭代次数。但当增大迭代次数仍然不收敛就需要对数据进行异常值检验等进一步处理。通常把一些表达量极低或者极高的基因删除掉,这个问题就可以解决。ImpulseDE2
plotHeatmap 函数,可以借助 ComplexHeatmap 对数据整体进行热图的绘制同时提取不同类的基因,也可以使用plotGenes看某一个基因的表达情况。EBSeq-HMM
Up-Up-Down-Down-Down 之类的若干 path,然后你可以选出你感兴趣的 path 进行后续分析。后续感受
参考资料
http://a-little-book-of-r-for-biomedical-statistics.readthedocs.io/en/latest/
https://laranikalranalytics.blogspot.com/2018/07/time-series-analysis-with-documentation.html
https://www.displayr.com/smoothing-time-series-data/?utm_medium=Feed&utm_source=Syndication
https://www.analyticsvidhya.com/blog/2015/12/complete-tutorial-time-series-modeling/
· 分享链接 https://kaopubear.top/blog/2018-08-25-rnaseqtimedata/