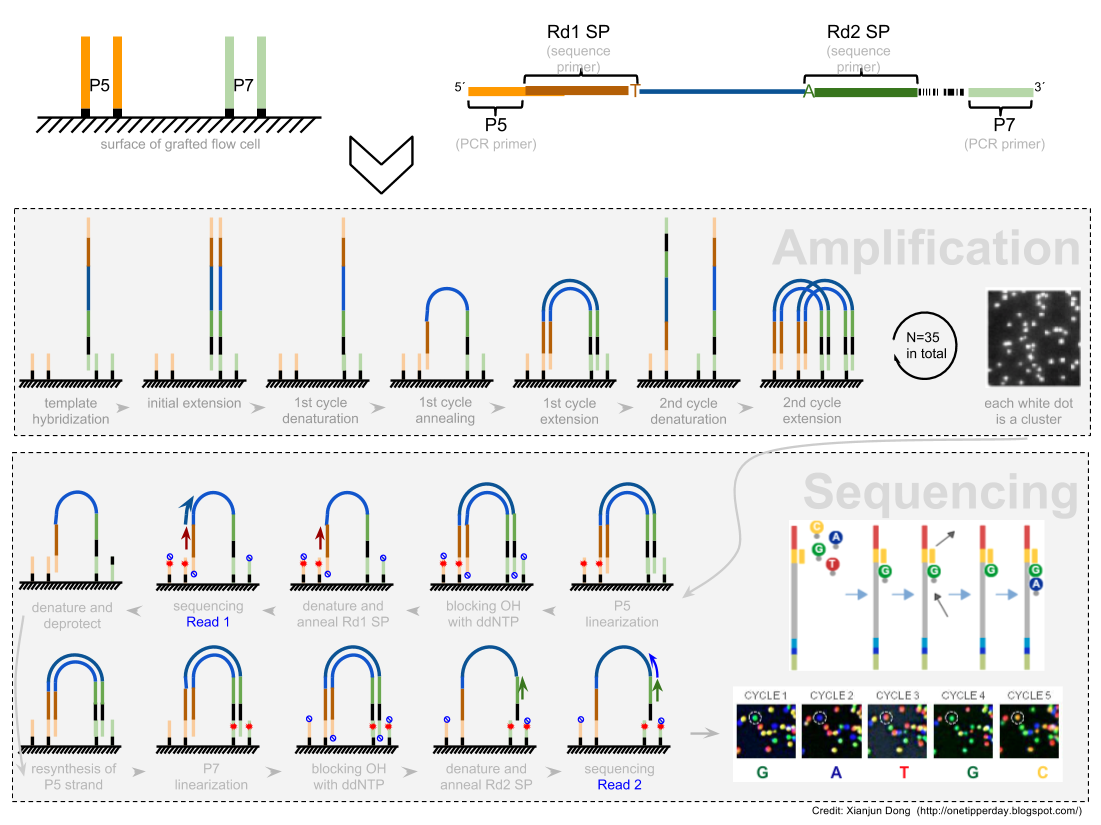
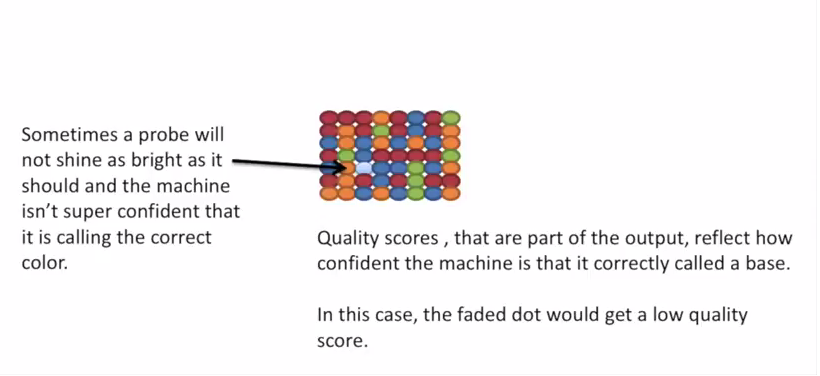
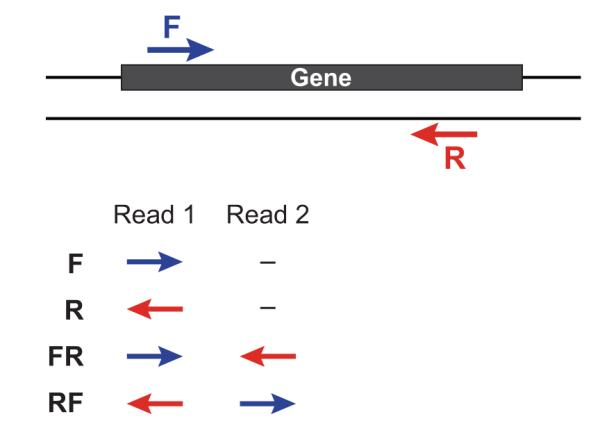
2021 年 12 月更新,距离写这篇文章已经过去 5 年了,但是今天发现这篇文章还会有人看。感慨一下。以及如果你只是想通过比对后的 bam 文件判断一下链特异性建库与否,这个文章太长了,直接去用RSeQC这个软件的 本文最早写于 2017 年,时间略显久远,各位选择性吸收 关于链特异性测序的若干问题,很久以前就以为自己想清楚了,但是每次提起它的时候又容易重新产生各种各样的小困惑。于是整理一下,以免以后再时不时犯迷糊。很多东西就是这样,你以为的明白并不是真的明白,一年前的明白和一年后的明白也不是同一个明白。我这么说,不知道你能明白还是不明白。 下图是一个大概的 RNA-seq 基本流程 把 RNA 破碎成小片段,然后将 RNA 转变成一条 cDNA,这一步需要用到反转录酶 reverse transcriptase (RT) 才能用 RNA 作为模板合成 DNA。 不论是转录还是反转录都需要引物。通常如果我们要 mRNA,那就可以用 oligo-dT 作为 RT 的引物,但是用它有两个问题,第一个是只能反转录那些有 A 尾巴的 RNA,第二个问题是 RT 不是一个高度持续性的聚合酶,可能让转录提前发生终止,造成的结果就是 3'端要比 5'端 reads 富集,这样就会使得后续定量分析带来 bias。 另一种常用的引物称为随机引物,随机引物的好处是没有 A 尾巴的诸如 ncRNA 也被留下了,而且不会存在明显的 3'端偏差。但是很多研究也发现,所谓的随机引物根本就不随机,这也是测序结果中,通常前 6 个碱基的 GC 含量分布特别不均匀的原因。这几个碱基 GC 含量均匀很可能不是接头或者 barcode 那些东西,其实是 Illumina 测序 RT 这一步的 random hexamer priming 造成的 bias,很多人在处理数据的时候会把这几个碱基去掉,其实很多时候真多 RNA-seq 数据去不去掉基本什么影响,不过开头如果有低质量的碱基倒是应该去掉。 随后是第二条链合成,这一步用是 DNA 聚合酶,以刚才和成的第一条链作为模板。 接下来就是在序列两端加上接头,加接头一方面是为了让机器可以识别这些序列,把这些序列固定;二是为了让多个样品可以同时上机,平摊每个样品的测序价格。双端测序为了让 read 从两边开始延伸,也需要在两端有所需的引物。 所谓双端测序,因为很多时候 read 的长度要短于 insert,为了增加覆盖度于是就想出了从 insert 两端同时测序的办法。使得测序深度增加的同时也能够用来判断 isoform 方向。 对于 illumina 数据,有一条 5-3 的 universal adaptor;还有一条是 3-5 的 indexed adatpor,这条引物含有特意的 barcode。需要说明的是,在双端测序中,如果 insert 不是足够长,那么 R1 可能就会测到 R2 的引物,同时 R2 可能会测到 R1 引物的反向互补序列。 大概的意思就是下面两张图。 加了接头以后进行 PCR 的扩增。扩增后就开始测序,测序的过程如下图所示。 测序的基本思想是机器识别四种碱基发出的不同颜色的荧光,可以理解为一个 flow cell 立着非常多序列,机器一层一层扫过去,通过识别荧光而判断这一层每个序列的碱基是什么。 因为一个 cell 密密麻麻的全是荧光信号,机器并不是总能把每一个判断的非常准确,如果某一个荧光信号没有那么清晰,这个碱基的测序质量就比较低,如下图。 有的时候,如果一大片点都是同一种荧光,机器也可能犯晕,不知道到底哪一个荧光属于哪一个序列。这种情况尤其是在序列的前几个碱基容易发生。 The sequencing machine uses the first few bases to establish where the cDNA fragments are on the flow cell. If all of the bases in one part of the flow cell are all the same, like 'C', and all show up green in the picture, then the colors will bleed together and it will not be clear where exactly all of the reads are. In contrast, if you have a lot of different colors in a region, it's easier to determine where each one is, even with a little color bleed. 和普通的 RNAseq 不同,链特异性测序可以保留最初产生 RNA 的方向,普通建库方式为什么不行呢?因为传统建库方式通过两个接头的 ligation 把 RNA 已经变成了双链 DNA,最后的文库中一部被测序的链对应正义链(sense strand),一部分被测序的链测是反义链。 链特异性建库方式有不止一种,对应到不同的软件又有不同的叫法,下面是几种称呼。要记住的是 dUTP 测序方式的名字是 fr-firstrand,也是 RF。 至于具体的 read 方向接下来通过更详细的 IGV 截图说明问题。 链特异性建库方式(以目前最常用的 dUTP 为例,如下图所示)首先利用随机引物合成 RNA 的一条 cDNA 链,在合成第二条链的时候用 dUTP 代替 dTTP,加 adaptor 后用 UDGase 处理,将有 U 的第二条 cDNA 降解掉。 这样最后的 insert DNA fragment 都是来自于第一条 cDNA,也就是 dUTP 叫 fr-firststrand 的原因。对于 dUTP 数据,tophat的参数应该为 说到链特异性测序,实在让人困惑的是各种链的概念,尤其是翻译成中文就更说不清了。 DNA 的正链和负链,就是那两条反向互补的链。参考基因组给出的那个链就是所谓的正链(forword),另一条链是反链(reverse)。但是这正反一定不能和正义链(sense strand)反义链(antisense strand)混淆,两条互补的 DNA 链其中一条携带编码蛋白质信息的链称为正义链,另一条与之互补的称为反义链。但是携带编码信息的正义链不是模板,只是因为它的序列和 RNA 相同,正义链也是编码链。而反义链虽然和 RNA 反向互补,但它可是真正给 RNA 当模板的链,因此反义链也是模板链。 总结两点 正义链(sense strand)= 编码链(coding strand)= 非模板链 forword strand 上可以同时有 sense strand 和 antisense strand。因为这完全是两个不同的概念。 写这篇文章的原因,就是因为有人问我,链特异性测序数据 htseq-count 的结果是不是应该把正负链的基因分别在-s yes 和-s reverse 两个参数结果中统计出来再做下游分析。这里犯的错误就是我们混淆了基因组正反链和基因正义反义链的概念。 从前文的一个图我们可以总结出dUTP 方式测序 R1 文件中 read1 的方向和基因的方向(正义链)是相反的,而 R2 文件中的 read2 方向和基因的方向是相同的。 可以参考下面的两个 igv 文件 bam 截图。 首先解释一下 igv 两个颜色参数的意义 下面这个图示按照 igv 颜色选项中的 read strand 方向进行区分,可以看到所有红色 read 都是在正链方向(注意正链不是正义链),而所有蓝色的 read 都是负链方向。下面基因的方向是正链方向,也就是和粉色的 read 同向的,如果你把鼠标放到随意一个粉色的 read 上,就能看到显示的信息是second of pair,也就是pair 中的 read2(R2);反之如果你在蓝色的 read 上面,就会显示信息是 first of pair,也就是 R1 。 总结,dUTP 测序中 pair read 中的 read1(R1)和基因方向相反,read2(R2)和基因方向相同 再看下面这张图 这张图展示了两个基因 1 和 2,我们可以发现gene1 的正义链就在正链上,而gene2 的正义链其实是在反链上。看 read 情况,a,c 两个 read 虽然针对正链负链而言方向一致,都是负链方向,但是如果把a 是 pair 中的 read1(first of pair ),而c 是 pair 中的 read2(second of pair)。也就是说,read 方向一致,但一个是 read1 一个是 read2,说明这两个 read 对应的基因一定是反向的。同样的道理,虽然b,d 都是两个方向为负链的 read,但是 b 其实是所在 pair 的 read2(second of pair),而 d 是所在 pair 的 read1(first of pair)。 再次强调,dUTP 测序中 pair read 中的 read1(R1)和基因方向相反,read2 和基因方向相同 当使用 read strand 来进行颜色区分的时候,每一个基因上两种颜色的分布应该相对均匀(也就是所谓的 pair end)。 如果这个时候把颜色选项改为按照 geng1 的 read 全部变成了紫色,而 gene2 的 read 全部变成了粉色。 如果是非链特异性测序,在 再一次总结: STAR mpping 时无需特别设置,但如果不是链特异性数据且下游分析要用到 cufflinks 则需要增加参数 --outSAMstrandField intronMotif。为的是增加一个 XS 标签。 If you have stranded RNA-seq data, you do not need to use any specific STAR options. Instead, you need to run Cufflinks with the library option --library-type options. For example, cufflinks... --library-type fr-firststrand should be used for the standard dUTP protocol, including Illumina’s stranded Tru-Seq. hisat2 --rna-strandness RF 目的也是给比对序列添加一个 XS 标签以区分方向,方面 cufflinks 使用。 For single-end reads, use F or R. 'F' means a read corresponds to a transcript. 'R' means a read corresponds to the reverse complemented counterpart of a transcript. For paired-end reads, use either FR or RF. tophat --library-type option fr-firststrand 具体解释参见下表 htseq-count -s reverse/yes(看反义链) For RSEM --forward-prob 0(正义链)1(看反义链) The RNA-Seq protocol used to generate the reads is strand specific, i.e., all (upstream) reads are derived from the forward strand. This option is equivalent to --forward-prob=1.0. With this option set, if RSEM runs the Bowtie/Bowtie 2 aligner, the '--norc' Bowtie/Bowtie 2 option will be used, which disables alignment to the reverse strand of transcripts. (Default: off) Probability of generating a read from the forward strand of a transcript. Set to 1 for a strand-specific protocol where all (upstream) reads are derived from the forward strand, 0 for a strand-specific protocol where all (upstream) read are derived from the reverse strand, or 0.5 for a non-strand-specific protocol. (Default: 0.5) sXpress --rf-stranded / --fr-stranded(看反义链) --fr eXpress only accepts alignments (single-end or paired) where the first (or only) read is aligned to the forward target sequence and the second read is aligned to the reverse-complemented target sequence. In directional sequencing, this is equivalent to second-strand only. --rf eXpress only accepts alignments (single-end or paired) where the first (or only) read is aligned to the reverse-completemented target sequence and the second read is aligned to the forward target sequence. In directional sequencing, this is equivalent to first-strand only. trinity --SS_lib_type RF Trinity performs best with strand-specific data, in which case sense and antisense transcripts can be resolved. RF: first read (/1) of fragment pair is sequenced as anti-sense (reverse(R)), and second read (/2) is in the sense strand (forward(F)); typical of the dUTP/UDG sequencing method. 到这里,会想问两个问题。有时候我们不知道数据的建库方式是不是链特异性的,如果弄错了结果会怎么样呢? 如果你用 STAR mapping 完可以用 igv 像上文提到的那样,看看是不是链特异性测序。 下面是两个真是数据的 count 统计情况。 对于仅仅进行基因表达定量来说,把链特异性数据当作普通建库数据来处理,可以观察第 2 列数据和第 4 列数据。具体某一个基因而言,影响不会太大,因为绝大多数反义链本身表达量就非常低。 但是因为区分了方向,又会使得 noFeature 的数目更多一些。不过两者总体影响不会差别非常大。如果不能判断建库方式,在 htseq 中使用-s no 参数是一个比较保险(虽然不是非常精确)的做法。 相反,如果把普通建库方式的数据当作链特异性数据来处理。 比如在 htseq-count 中使用了-s reverse 参数,这个时候只有 R2 方向和基因方向相同的 pair 才用来算作一个 count,所有 R2 和基因方向不同的 pair 就被当作 no feature 了。这样的结果影响可以通过下面的表格观察。 用正常方法数出的 noFeature 是 6 万左右,而用-s yes 或者 reverse 数出来的 noFeature 就接近 46 万了。将近 40 万的 read 被丢掉。 所以,如果把普通建库的数据误当作链特异性数据来处理极有可能会损失大量的数据,如果弄错了链特异性建库的方式,那可能你就没几个 read 剩下了。另外,计算出来的结果自然也会有非常大的差异,是不准确的。 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。
infer_experiment.py命令跑一下就好。祝好~
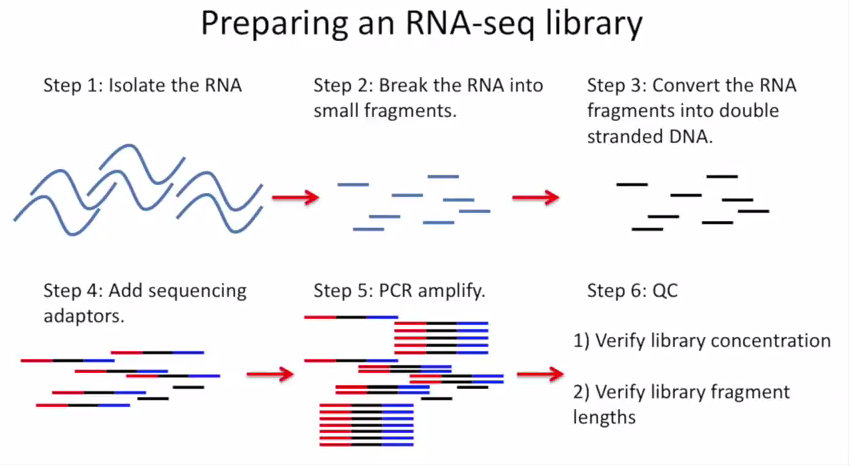
RNA-seq 基本流程
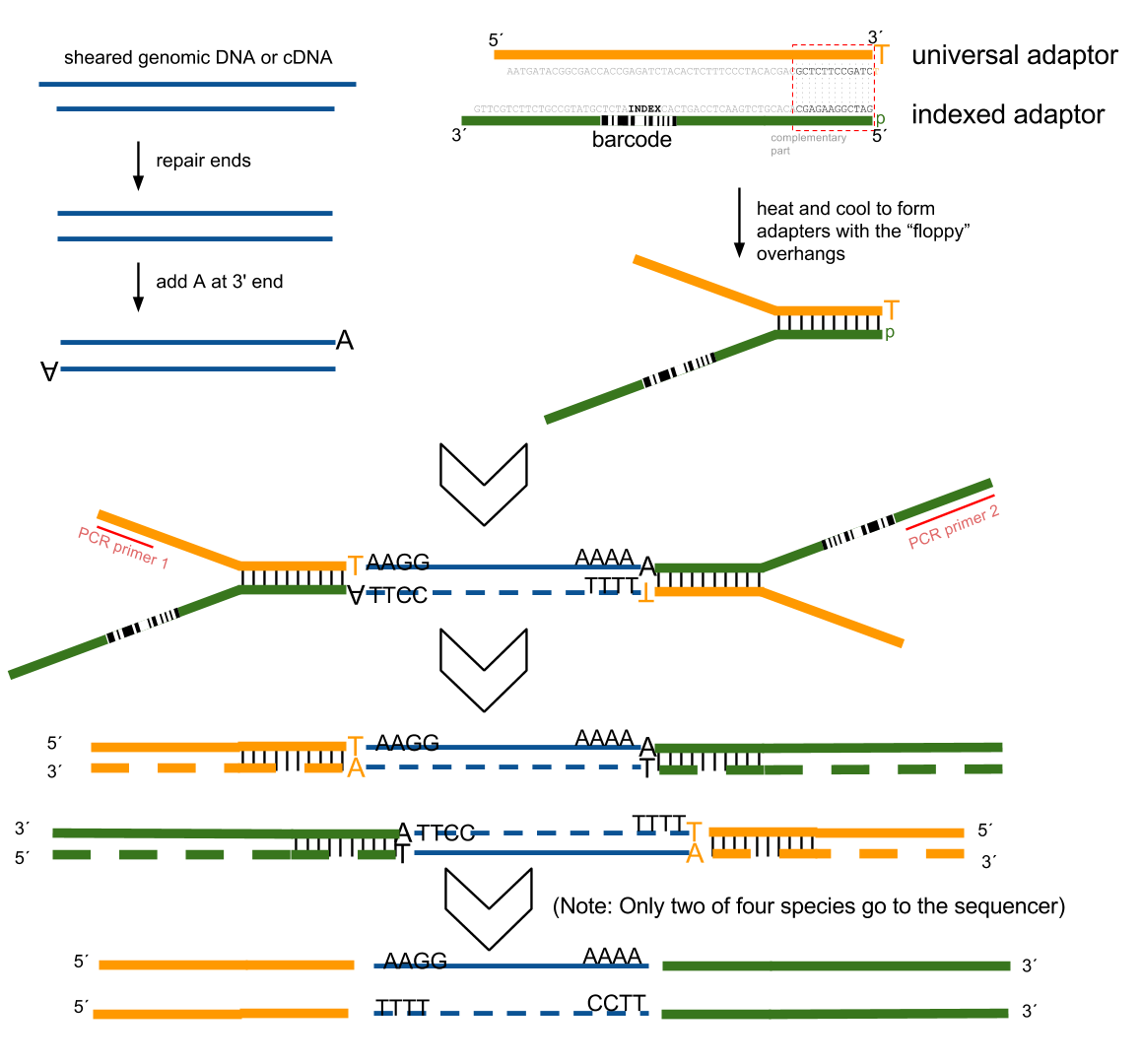
Adapter element
Requirement
Location
Function
Amplification element
Required
5′ and 3′ terminus
Clonal amplification of the construct
Primary sequencing priming site
Required
Adjacent to the insert
Initiating the primary sequencing reaction
Barcode/Index
Optional
5′-end of the insert/Between the sequencing priming site and its respective amplification element
Provides a unique label to sequences from different samples. Allows pooling of multiple experiments in a single sequencing reaction.
Paired-end sequencing priming site
Optional
Adjacent to the insert on the side opposite of the primary sequencing priming site
Sequencing into the insert on the end opposite of the primary read
Index sequencing priming site
Optional
Complementary to the 5′-end of the sequencing priming site
Sequencing of the index

链特异性测序
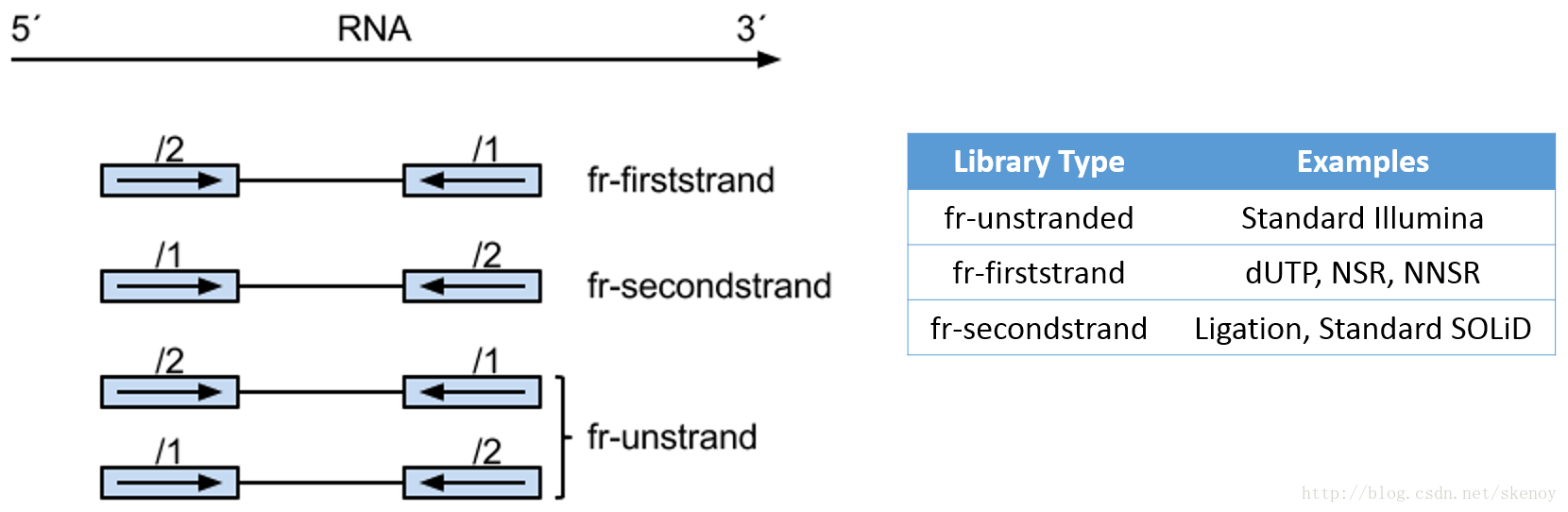
–library-type fr-firststrand。这里的 first-strand cDNA 可不是 RNA strand,在使用htseq-count 时,真正的正义链应该是使用参数-s reverse 得到的结果。正正反反不清楚
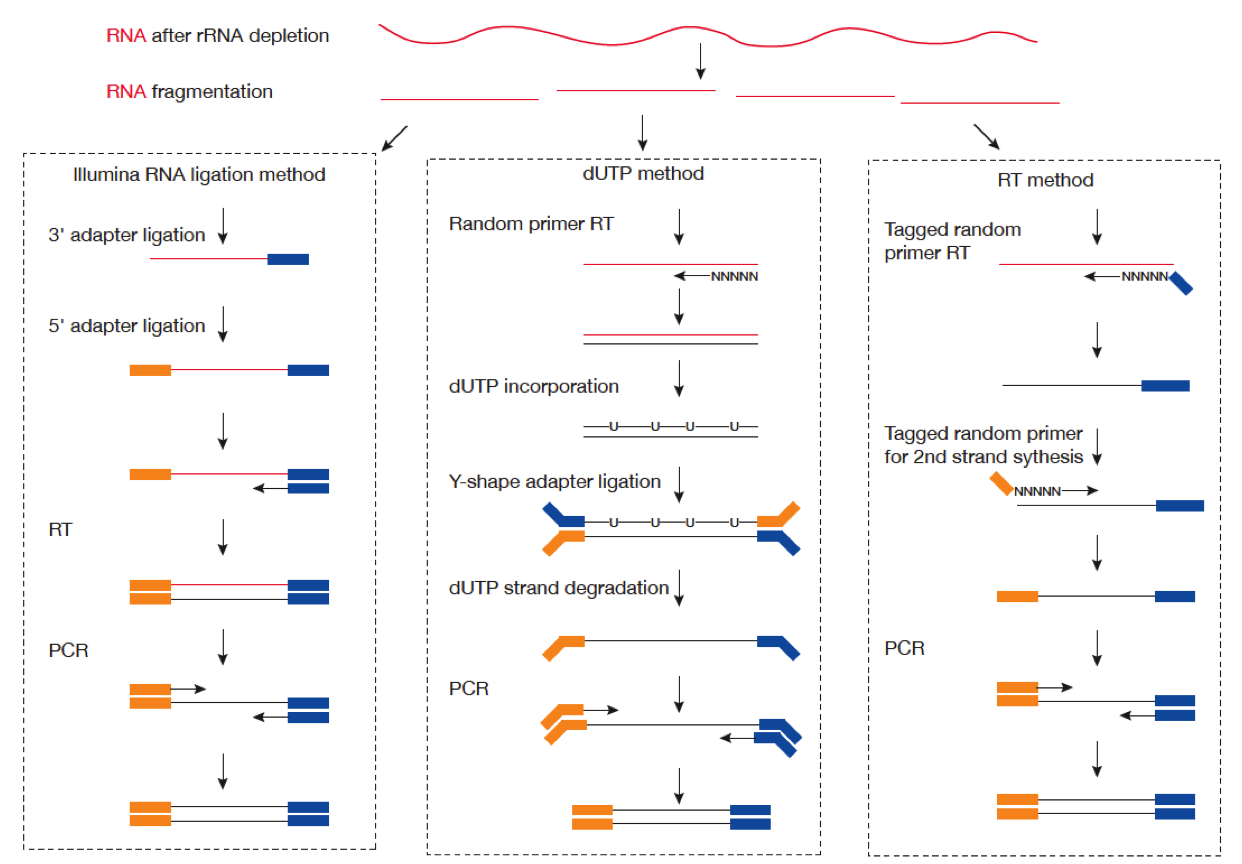
dUTP 到底是怎么回事
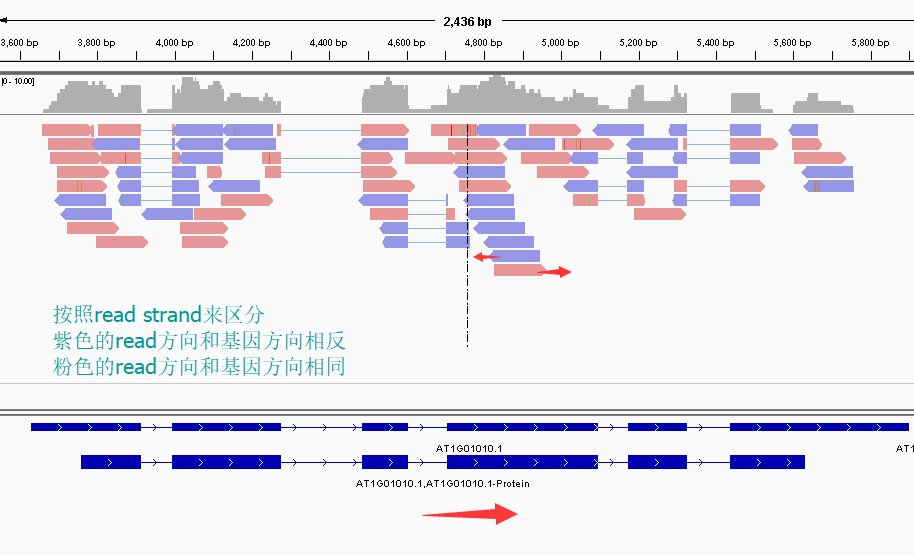
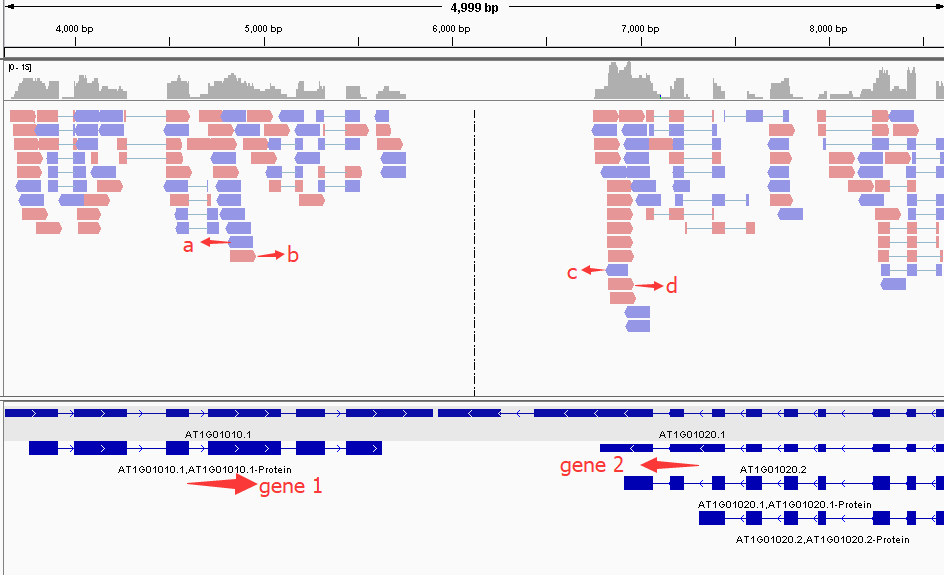
first of pair of strand来区分,会出现下图的变化。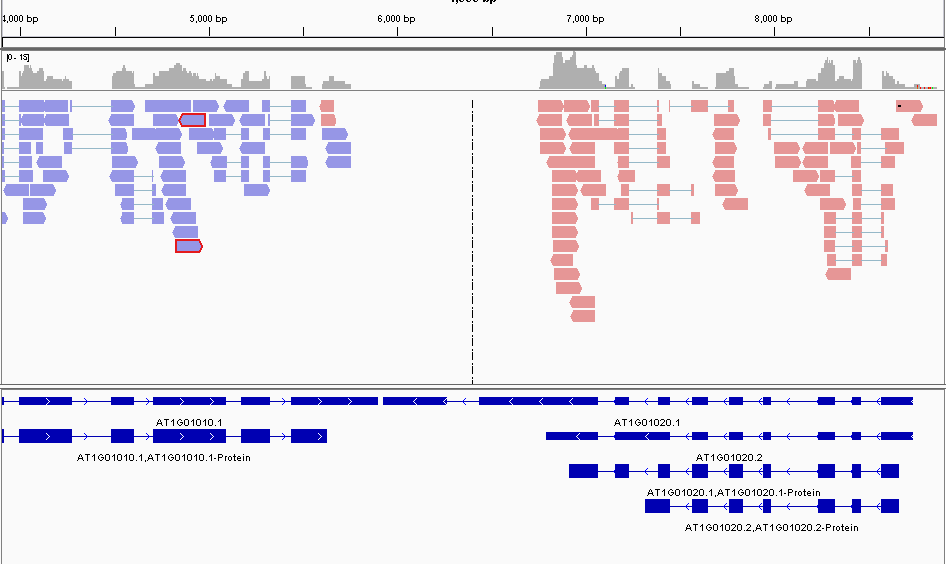
first of pair of strand模式下,同一个 gene 相关的 read 颜色也是明显混杂的。如下图\
几个常用软件的设置
With this option being used, every read alignment will have an XS attribute tag: '+' means a read belongs to a transcript on '+' strand of genome. '-' means a read belongs to a transcript on '-' strand of genome.
Library Type
Examples
Description
fr-unstranded
Standard Illumina
Reads from the left-most end of the fragment (in transcript coordinates) map to the transcript strand, and the right-most end maps to the opposite strand.
fr-firststrand
dUTP, NSR, NNSR
Same as above except we enforce the rule that the right-most end of the fragment (in transcript coordinates) is the first sequenced (or only sequenced for single-end reads). Equivalently, it is assumed that only the strand generated during first strand synthesis is sequenced.
fr-secondstrand
Ligation, Standard SOLiD
Same as above except we enforce the rule that the left-most end of the fragment (in transcript coordinates) is the first sequenced (or only sequenced for single-end reads). Equivalently, it is assumed that only the strand generated during second strand synthesis is sequenced.
stranded=no, a read is considered overlapping with a feature regardless of whether it is mapped to the same or the opposite strand as the feature. For stranded=yes and single-end reads, the read has to be mapped to the same strand as the feature. For paired-end reads, the first read has to be on the same strand and the second read on the opposite strand. For stranded=reverse, these rules are reversed.
参数错了又怎样?
不过可以注意 noFeature 和 ambiguous 这两个值,因为基因组中存在两个基因分别在正链和负链且又重叠的情况,不区分方向会比区分方向的 ambiguous 数目多一些。因为如果不能通过方向来区分到底属于哪个基因,这样的 read 就会被认为是 ambiguous。
-s no
-s yes
-s reverse
N_noFeature
1290001
16837194
1480658
N_ambiguous
633021
16413
74710
AT1G01010
58
2
56
AT1G01020
65
0
65
AT1G03987
5
5
0
AT1G01030
296
5
291
AT1G01040
901
5
1078
AT1G03993
0
182
0
AT1G01050
428
0
434
AT1G03997
1
7
0
AT1G01060
85
0
85
AT1G01070
73
0
73
AT1G04003
0
0
0
AT1G01080
1166
15
1151
AT1G01090
2901
0
2901
AT1G01100
1560
0
1560
AT1G01110
82
0
82
AT1G01120
484
0
484
AT1G01130
72
9
63
AT1G01140
518
3
515
AT1G01150
0
1
0
AT1G01160
356
192
551
AT1G04007
4
189
0
AT1G01170
55
11
423
-s no
-s yes
-s reverse
N_unmapped
729831
729831
729831
N_multimapping
443861
443861
443861
N_noFeature
63787
4591673
4599916
N_ambiguous
594720
27992
27789
AT1G01010
101
54
47
AT1G01020
84
41
43
AT1G03987
1
0
1
AT1G01030
80
37
43
AT1G01040
499
279
293
AT1G03993
0
33
25
AT1G01050
634
312
337
AT1G03997
0
0
0
AT1G01060
3274
1644
1630
AT1G01070
97
43
54
AT1G04003
0
0
0
AT1G01080
585
303
282
AT1G01090
1768
907
861
AT1G01100
1132
549
583
AT1G01110
60
21
39
AT1G01120
1099
573
526
· 分享链接 https://kaopubear.top/blog/2017-11-11-ssrna/