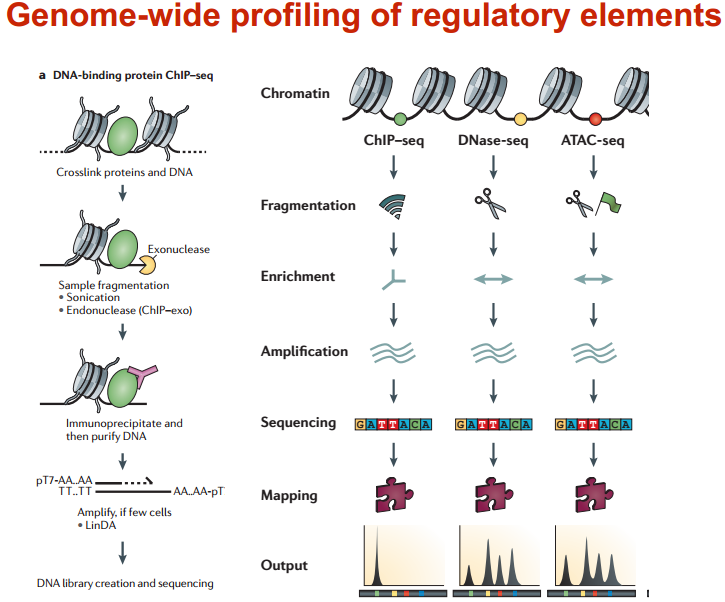
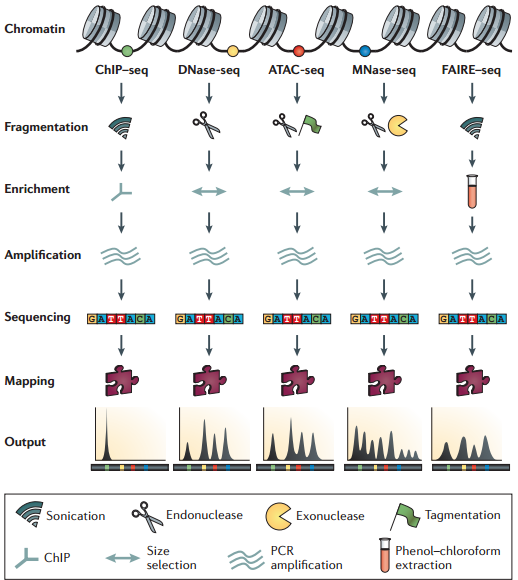
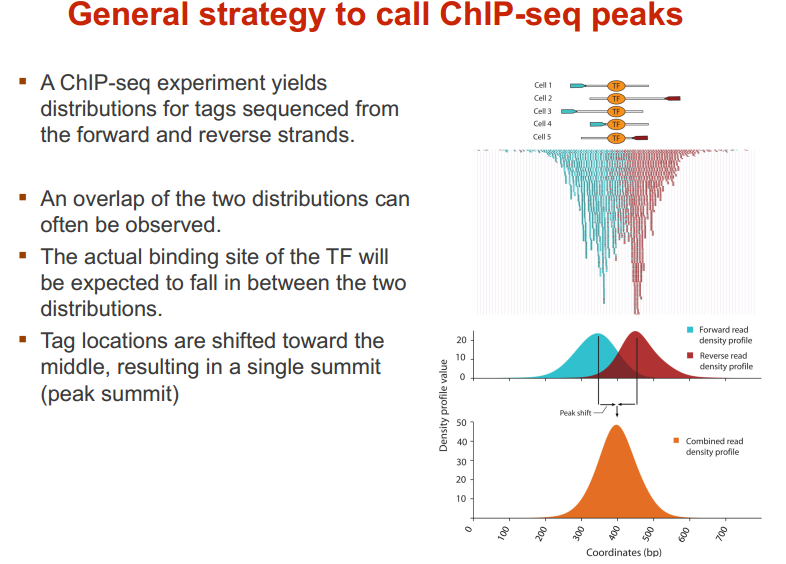
在人中,有 300 个 TF 结合在核心启动子区域;有 1500 个结合在基因其他区域,可以调节一系列基因 图示 ChIP-seq: DNase-seq DNase I 超敏感位点(DHS)是对 DNaseⅠ 高度敏感的活性染色质区域,DNase 测序(DNase-seq)是进行全基因组 DHS 分析的常用方法 DNase I 是一种非特异性核酸内切酶,基于它们对切割的过敏性,长期以来就被用于对“开放”染色质位点的作图 chromatin open 的位置很容易有其他蛋白的结合 由于多种蛋白质可以与相同序列相结合,有必要整合 DNase-seq 测序数据和 ChIP-seq 测序数据来对引起特定 DNase 足迹的蛋白质进行定性鉴定 不依赖于抗体或表位标签,DNase-seq 可以用来在一次实验中分析大量蛋白质的基因组分布 从大范围来看,结合的位置凸起。如果从小范围来看,空着的位置刚好是一个可能的 motif ATAC-seq (Assay for transposase- accessiblechromatin using sequencing) 文章原图 A genomic locus analysed by complementary chromatinprofiling experiments reveals different aspects ofchromatin structure ChIP–seq reveals binding sites of specifictranscription factors (TFs); DNase-seq, ATAC-seq andFAIRE–seq reveal regions of open chromatin; andMNase-seq identifies well-positioned nucleosomes. In ChIP–seq, specific antibodies are used toextract DNA fragments that are bound to the targetprotein, either directly or through other proteins ina complex that contains the target factor. In DNase-seq, chromatin is lightly digested by theDNase I endonuclease. Size selection is used toenrich for fragments that are produced in regions ofchromatin where the DNA is highly sensitive to DNaseI attack. ATAC-seq is an alternative method to DNase-seq thatuses an engineered Tn5 transposase to cleave DNA andto integrate primer DNA sequences into the cleavedgenomic DNA (that is, tagmentation). Micrococcal nuclease (MNase) is an endo–exonucleasethat processively digests DNA until an obstruction,such as a nucleosome, is reached. In FAIRE–seq, formaldehyde is used to crosslink chromatin, and phenol–chloroform is used to isolate sheared DNA. Some TFs almost always bind in proximal promoter regions Others bind to many regions Position weight matrix (PWM) 把所有碱基出现的次数相加,高度表示可信度 这种方法过于简单,不能表示出碱基之间的关系。 假设各个碱基之间均为独立 Given a collection of genes that are likely to be regulated by the same TFs (or orthologous genes across different species — methods based on phylogenetic footprinting principles), find the TF-binding motifs in common 但是问题是不知道 motif 是什么,找不到相关的基因,而且如何排除背景干扰 比较保守的非编码区域可能有 Expectation-Maximization In each iteration, it learns the PWM model and identifies examples of the matrix (sites in the input sequences) 在每一次迭代中,学习一个 PWMmodel 然后再通过输入的序列进行比对 MEME works by iteratively refining PWMs and identifying sites for each PWM(不同的迭代直到找到一个最合适的 PWM) The intuitive idea is as follows: Start with a k-mer seed (random or specified) 通常是 6 个 For every k-mer in the input sequences, identify its probability given the PWM model 计算 k-mer 在输入序列中给出 PWM 出现的概率 Calculate a new PWM, based on the weighted frequencies of all k-mers in the input sequences 例子 1 1.1 1.2 1.3 首先设置 model, 然后经历 Estep 和 Mstep, 找到合适的 PWM 然后将 PWM 进行极大似然转换并取 log 然后看输入序列中出现该 motif 的概率 人的大多数结合位点都是在内含子和基因间区 Stronger sites are not closer to differentially regulated genes (not necessarily more functional) Majority of functional sites not conserved 目前很难预测靶基因 核心思想 TF 在基因组上的结合其实是一个随机过程,基因组的每个位置其实都有机会结合某个 TF,只是概率不一样 peak 出现的位置,是 TF 结合的热点,而 peak-calling 就是为了找到这些热点。 热点:位置多次被测得的 read 所覆盖(我们测的是一个细胞群体,read 出现次数多,说明该位置被 TF 结合的几率大)。 read 出现多少次算多:假设 TF 在基因组上的分布没有任何规律,测序得到的 read 在基因组上的分布也必然是随机的,某个碱基上覆盖的 read 的数目应该服从二项分布。 当 n 很大,p 很小时,二项分布可以近似用泊松分布替代 是泊松分布唯一的参数,n 是测序得到的 read 总数目,l 是单个 read 的长度,s 是基因组的大小。 我们可以算出在某个置信概率(如 0.00001)下,随机情况下,某个碱基上可以覆盖的 read 的数目的最小值,当实际观察到的 read 数目超过这个值(单侧检验)时,我们认为该碱基是 TF 的一个结合热点。反过来,针对每一个 read 数目,我们也可以算出对应的置信概率 P。 实际情况由于测序、mapping 过程内在的偏好性,以及不同染色质间的差异性,相比全基因组,某些碱基可能内在地会被更多的 read 所覆盖,这种情况得到的很多 peak 可能都是假的。 MACS 考虑到了这一点,当对某个碱基进行假设检验时,MACS 只考虑该碱基附近的染色质区段(如 10k),此时,上述公式中 n 表示附近 10k 区间内的 read 数目,s 被置为 10k。当有对照组实验(Control,相比实验组,没有用抗体捕获 TF,或用了一个通用抗体)存在时,利用 Control 组的数据构建泊松分布,当没有 Control 时,利用实验组,稍大一点的局部区间(比如 50k)的数据构建泊松分布。 read 只是跟随着 TF 一起沉淀下来的 DNA fragment 的末端,read 的位置并不是真实的 TF 结合的位置。 在 peak-calling 之前,延伸 read 是必须的。不同 TF 大小不一样,对 read 延伸的长度也理应不同。 我们知道测得的 read 最终其实会近似地平均分配到正负链上,这样对于一个 TF 结合热点而言,read 在附近正负链上会近似地形成“双峰”。 MACS 会以某个 window size 扫描基因组,统计每个 window 里面 read 的富集程度,然后抽取(比如 1000 个)合适的(read 富集程度适中,过少,无法建立模型,过大,可能反映的只是某种偏好性)window 作样本,建立“双峰模型”。 最后,两个峰之间的距离就被认为是 TF 的长度 D,每个 read 将延伸 D/2 的长度 If we are given a set of ChIP-seq peaks, how to identify motif for the TF— use MEME To find out what the sequence motif resembles — use TomTom Use known motif to search peak regions — use FIMO Study common biological pathways or functions of potential target genes of the TF — use GREAT 刘晓乐实验室 ChIP-seq 数据分析流程 定义:包括一个有向无环图(DAG)和一个条件概率表集合。DAG 中每一个节点表示一个随机变量,可以是可直接观测变量或隐藏变量,而有向边表示随机变量间的条件依赖;条件概率表中的每一个元素对应 DAG 中唯一的节点,存储此节点对于其所有直接前驱节点的联合条件概率 性质:每一个节点在其直接前驱节点的值制定后,这个节点条件独立于其所有非直接前驱前辈节点 类似 Markov 过程,贝叶斯网络可以看做是 Markov 链的非线性扩展。这条特性的重要意义在于明确了贝叶斯网络可以方便计算联合概率分布。 通过基因表达来推测网络 每个节点不是一个基因合适若干基因 经典文章 主要过程 分析过程要给已经构建的相关性矩阵取逆 当样本很小时无法进行转换要使用 lasso 算法 关键在于如何确定公式中的 lamada 这样不需要所有节点之间都有边 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。cis-regulatory motifs
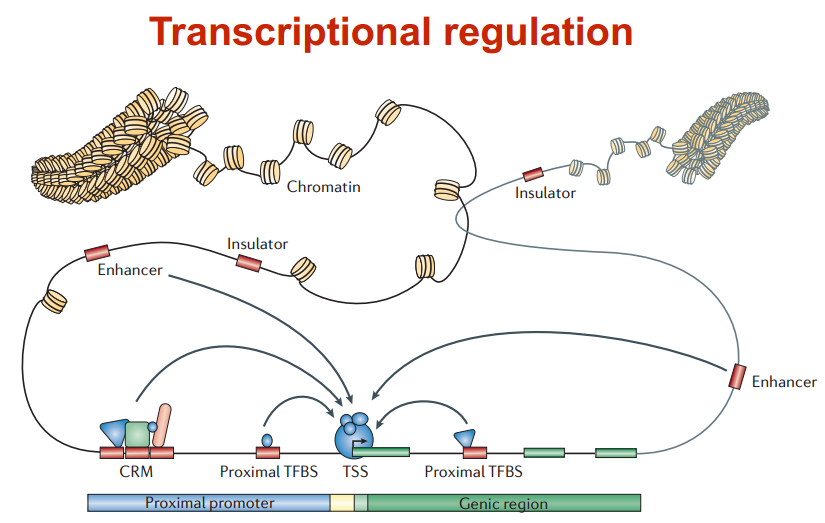
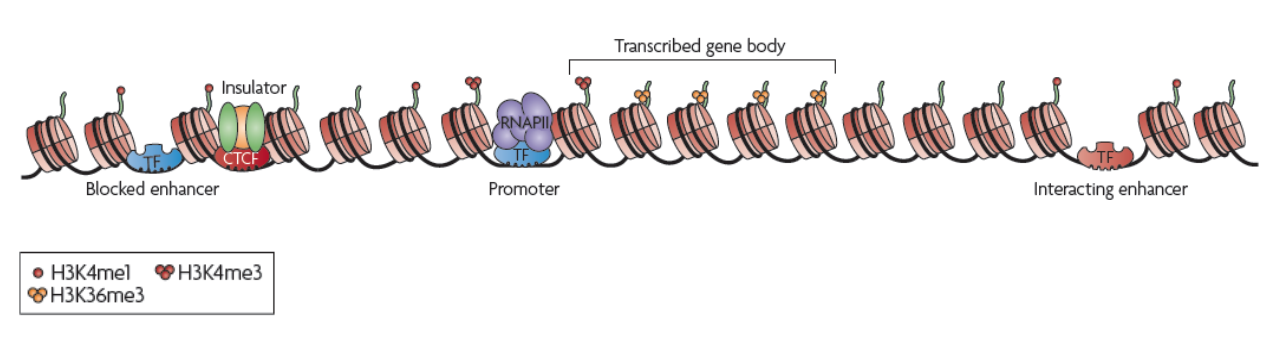
转录调控
全基因组研究调控原件的主要方法
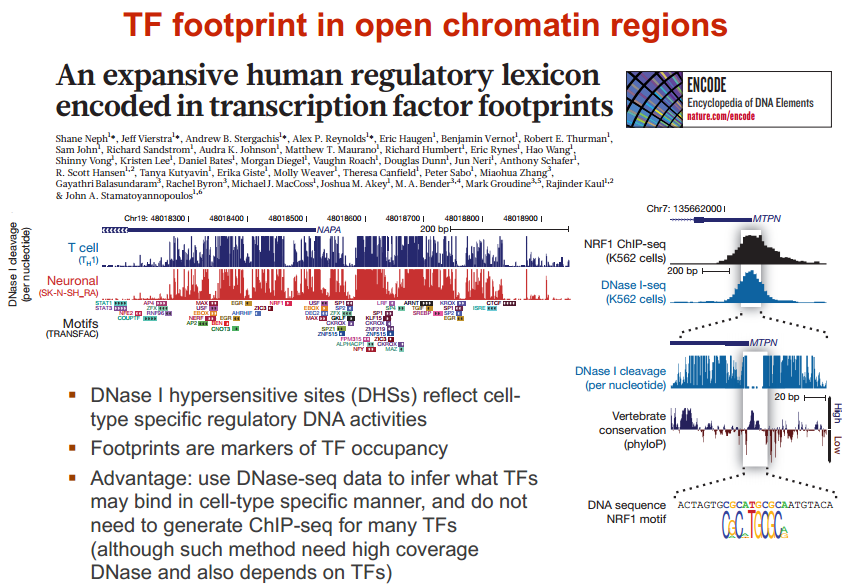
结合在哪里
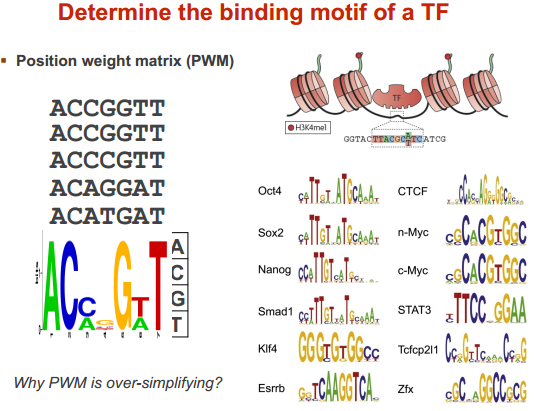
常用的表示方法
How is specificity of binding achieved
motif 定义

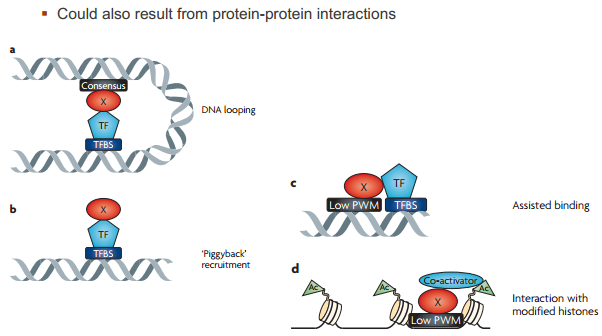
有时并非直接和 DNA 结合
How to identify TF binding sites?

没有序列比对的时候
最原始的方法是多重序列比对 MSA
PhyloCon — comparative genomic approach
Expectation-Maximization (EM) 目前最常用的方法(MEME)
Build a PWM by incorporating some of background frequencies 根据背景生成一个初始的 PWM
根据 input 序列中 k-mer 出现频率的权重更新 PWM




在 MEME 中
相关文献

过程


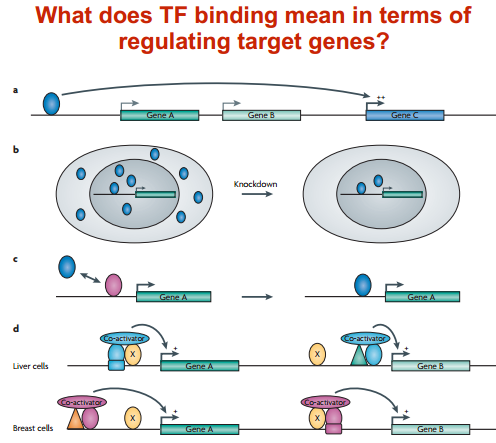
What does transcription factor binding mean in terms of regulating target genes?
ChIP-seq call peak 的策略和思想
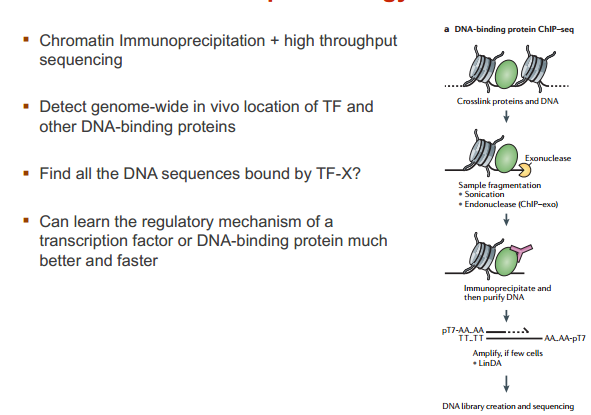
ChIP-seq 技术
macs 原理
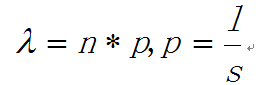

ChIP-seq 后续分析
基因调控网络
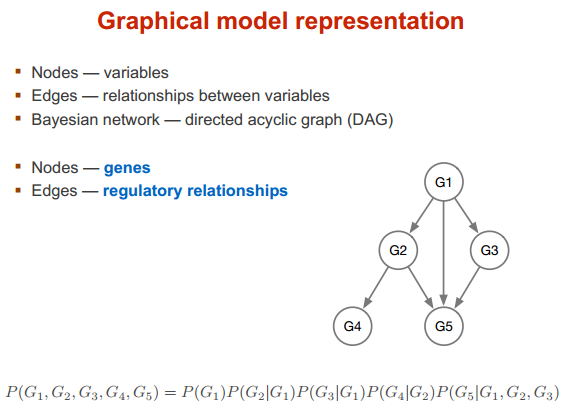
贝叶斯网络
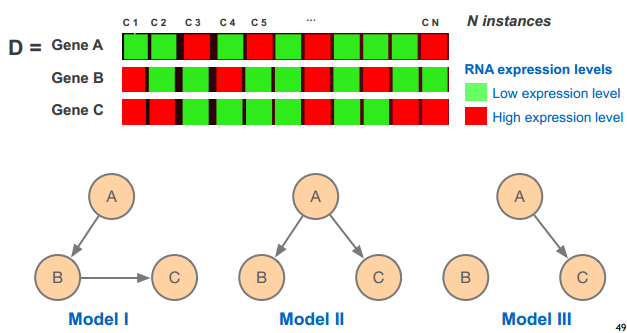
model 图形说明
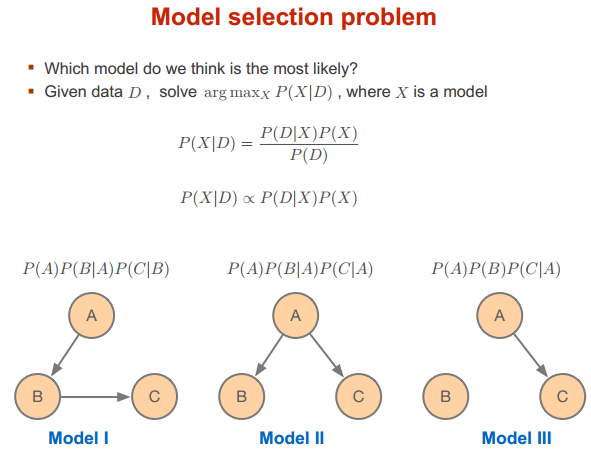
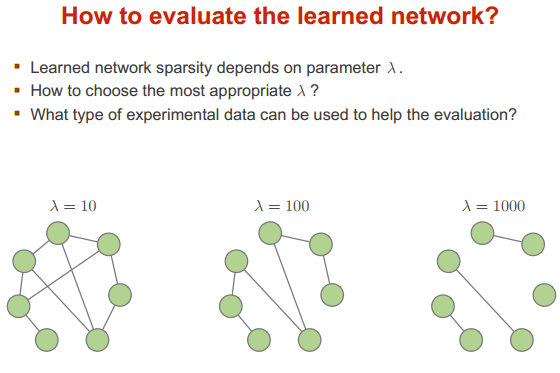
模型选择
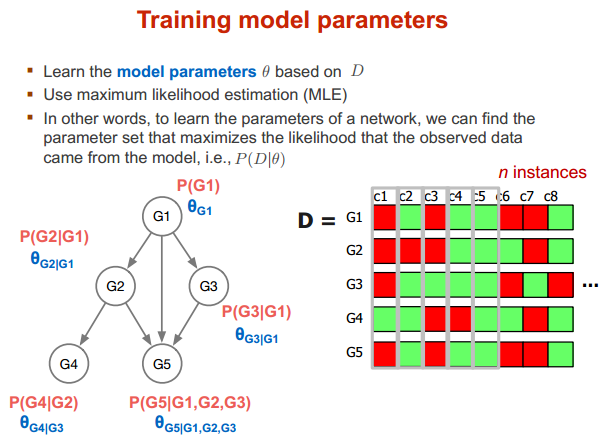
训练参数
CPD for discrete expression level

实际含义
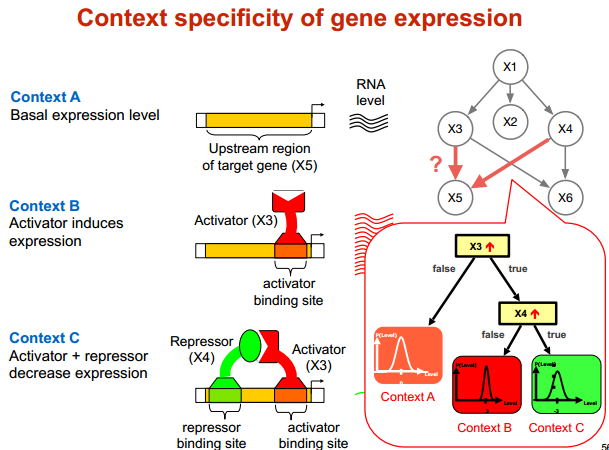
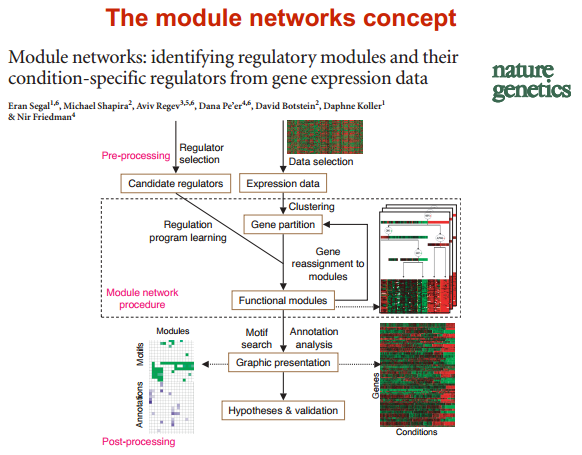
module network 模块网络

无方向图

· 分享链接 https://kaopubear.top/blog/2017-08-18-longxing-bioinfo-tf/