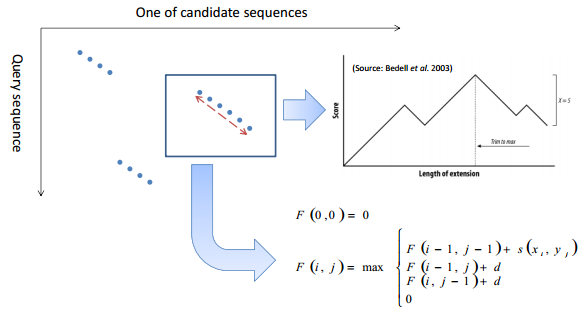
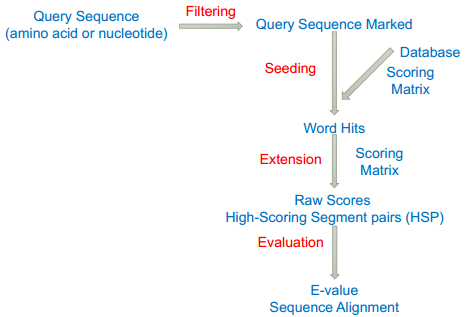
由芝加哥的 Needleman 和 Wunsch 两位于上个世纪 70 年代初提出,常被称之为 Needleman-Wunsch 算法。算法针对用户指定的打分函数,确定性地找出两条序列间的最优比对。 Needle-Wunsch 算法对两条序列所有残基进行全局比对的局限性。 1981 年,物理学家 Temple Smith 和数学家 Michael Waterman 在 Journal of Molcular Biology 上发表了一篇仅有四页的文章,提出 Smith-Waterman 局部比对算法 race back begins at the highest score inthe matrix and continues until you reach 0。And also the secondary best alignment 核心思想是给分数增加了下限 0 分 所有的回溯都是局部的,所有的最终比对也是局部的。引入止损下限,差异扩大之后重启比对,找到局部水平的相似性 Affine gap penalty 有限向量机 扩展公式 O(mn) 正比于 m*n 直系同源 ortholog 来自于不同的物种,演化过程中基因没有丢失,各物种中都有 chaining 旁系同源 paralog 来自于一个物种内部基因组的复制 netting Map the large numbers of short reads to a reference genome short: greater search sensitivity large: faster search speed BWA 和 bowtie 的相关算法,大大减少了对服务器的要求 如何快速的知道某段序列大约在基因组的哪个位置 核心是绕最后一个序列转圈 先给每一个 T 做 rotations, 再进行 sort, 生成 bw 矩阵,最后一列从头到尾就是 BWT 回溯 给每一个 T 的字母一个出现次数的排序,图示如下 类似于电话本中的索引结构 What if we need to check many queries We don't need to check every page of the phone book to find 'Ma' Sorting alphabetically lets us immediately skip 96% (25/26) of the book withoutany loss in accuracy Sorting the genome: Suffix Array (Manber & Myers1991) 所有具有相同 prefix(前缀)的 suffixes(后缀)会聚在一起,这样就可以进行类似于二分法的排除 全基因组建立 index An array of integers giving the startingpositions of suffixes of a string inlexicographical order 从中间的 index 开始找,过滤一半 效率 Total Runtime: O(m log n) Searching the array is very fast, but it takes time to construct Indexing-based local alignment Basic Local Alignment Search Tool 围绕最优比对路径进行计算 BLAST Ideas 核心思想:Seeding‐and‐extending Find matches (seed) between the query and subject 找到高度相似的小片段,种子 Extend seed into High Scoring Segment Pairs (HSPs) 向两端延伸并进行比对 Run Smith‐Waterman algorithm on the specified region only Assess the reliability of the alignment 打分评估 将序列切分,在数据库中定位候选序列和位置 得到候选序列和查询序列的 heatmap 去掉零散的 hit, 直留下对角线,形成 hit cluster 以 hit cluster 为基础向左右进行延伸直到分数不符合要去 在扩展的区域进行局部比对 blast 加速 分数评估,避免随机因素 E value 在随机情况下获得比当前分数高的可能比对条数,不是概率是个望值。p 为 0.05 时,E 也是 0.05。 BLAST 是一种启发式算法,不确定有最优解 只在有效区域应用动态规划算法 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。全局比对 Global Alignment
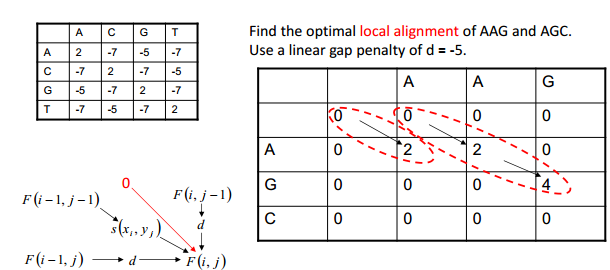
局部比对 Local Alignment
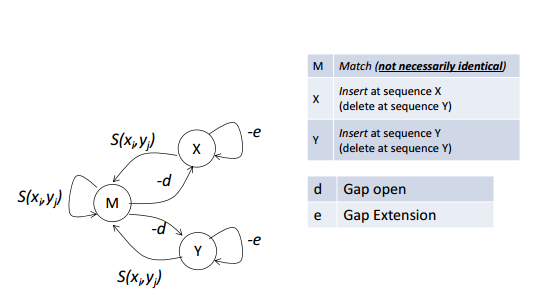
空位罚分的改进
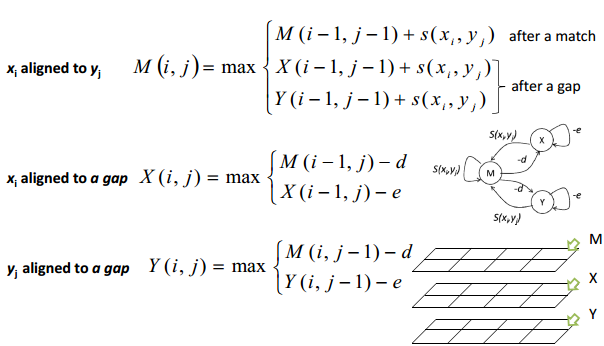
全局比对的时间复杂性
全基因组比对
同源 homology 的分类
NGS: Sequence alignment
In-exact alignment
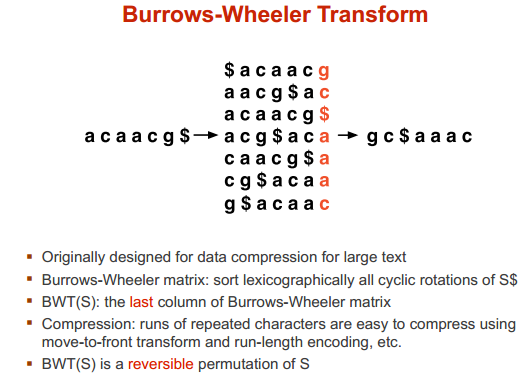
BWT 算法

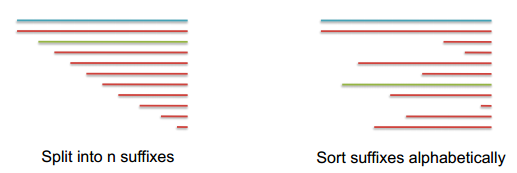
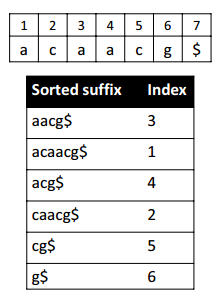
Suffix(后缀) Arrays

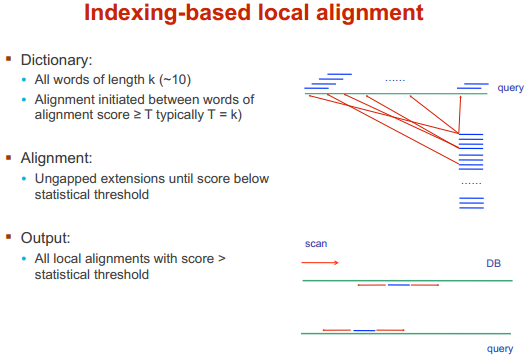
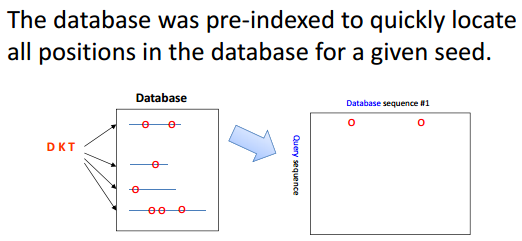
BLAST/Dot matrix

· 分享链接 https://kaopubear.top/blog/2017-08-14-longxing-bioinfo-blastmore/