Bioinformatics was like a box of chocolates. You never konw what you're gonna get ——阿飞正传 由于自己之前一直不喜欢用 cufflinks,所以后面的 stringtie 也是能不用就不用,偶尔用下也都是浅尝辄止。因为 stringtie 可以直接拿到基因的 TPM 值,比 RSEM 需要单独构建一次索引的操作省力些,所以最近自己注释了些基因就用它对十几个样本跑了一波基因定量的常规操作。心想做个表达矩阵进行下游分析,结果偶买噶,每个定量结果的行数(基因数)竟然都不一样。同一个注释文件定量出了不同的结果,检查一下原始基因数,发现有的定量结果行数要比实际基因数多 7 个,有的要多 5 个,还不尽相同。 遇事不怕事,先看看多出来了什么基因,我发现有八九个基因竟然会重复出现在定量结果中。回到注释文件里看这些基因为什么作妖,随便看了 3 个发现一个规律。这些基因对应的转录本 ID 并不是连续的(因为上游分析时我过滤掉了一些不符合自己要求的转录本),如下截图所示,gff 删掉了 2 和 3,结果在定量时出现了两个相同的 ID 。 如此这般,难道是 stringtie 不能识别不连续 ID 转录本的基因?暗自感觉这个 bug 无语的同时动手把那几个命名有 gap 的转录本 ID 重命名为连续,又测试了一次还是重复出现了这个问题。看来并不是 ID 不连续造成的,肯定还有一些背后的原因。 再一次仔细观察这些不连续的转录本信息, 好了,现在另一个问题是,如果是没有重叠区域的转录本会被按照独立的基因单独统计,那为什么不同的样本重复的基因数量并不一样呢?到底 stringtie 是怎么一个定量逻辑,到这里靠猜就解决不了问题了,只能去找原始文献和源代码。 历史的经验告诉我自己不太可能是第一个遇到这问题的人,于是问了一些朋友但也是答非所问。不如干脆去 GitHub 提个 issue,习惯性在提之前尝试搜了一下 我点进去发现内容还挺长,开发者和提问人有好几个回合的问答。读完不禁长处一口气,这个 issue 不光解答了我的困惑,还提供了一个如何提问的绝佳范本。非常值得我们去学习和反思。 以下是主要内容的摘录和简要评论: I am currently running stringtie on Arabidopsis rna-seq data sets and I am encountering duplication events that vary from sample to sample. The command I am running is: I am using the arabidopsis gtf downloaded from TAIR, and the reads I am running are downloaded from NCBI. My workflow is trimmomatic, hisat2, samtools_sort, samtools_index, stringtie. I am running stringtie v1.3.4d. My workflow was run on a llinux slurm cluster, and I have also verified the same results on my local machine (Ubuntu 18.04). I typically have 1 or 2 genes per run that have duplication events. A colleague running the same workflow on bovine data is having the same issue. 提问人在问题的描述中首先给出了自己处理的数据是什么物种,然后附上了自己处理的完整命令,另外也说了自己的参考基因组下载自哪里。同时写明了自己的分析流程和 stringtie 对应的软件版本。作者也强调自己在服务器集群和本地都是一个结果,以及自己的朋友也重复了这个问题——总有一两个基因在定量的时候会被重复呈现。 在开发者的第一次回复中提到了是不是注释文件做过某些处理,本身存在重复的 gene id,另外希望能提供一点有问题的 bam 文件,另外作者还非常贴心的提供了如何提取一些 bam 结果的方法。这里就不引用原始内容了。以下是提问者的第二次回复。 Sorry, the name of the gtf was confusing, it makes more sense in the context of our workflow. Yes, the gtf was produced from the file on the TAIR page named using the command from Cufflinks I cannot attach my file due to size constraints, but by downloading the above file and using that command you should be able to get the same reference file to work with. The file that results from the above command does not appear to have duplicates. In the output files from stringtie on my arabidopsis data, each file seems to have a random duplication of 1 of three genes. Those genes are: Here is a subset of my list of my output files (whole list is very long), and below each file name is what gene is duplicated (bash uniq command). The name of the file indicates the srx ID from NCBI. Each of these samples had multiple runs associated with it (sra). Before running through the pipeline these “sra” were combined into one large fastq file corresponding to their respective “srx”. You will notice that each file had different combinations of genes that were duplicated, but that they were always 1 of the 3 genes listed above. As I was thinking about it, I got paranoid that this combining of sra’s into srx may have caused the issue, so I redid it with a sample without combining (used sample SRR4426362). I got the same results. The attached files are a subset of the resulting *.bam that will cause a duplication event when used with this command and the above mentioned reference file: I used this command to see the duplicated gene in the gene abundance file: I have also included the output gene abundance file from this subset bam. I went through all of this again to verify that the results are still happening. Our workflow is using the latest editions of all of the other software as well. I had to rename the attached files with a ".txt" extension to be able to upload them to git hub, so I hope that the bam still work when you get it. 这一段回答内容比较长,提问者首先解释了自己对 gtf 文件进行了哪些操作以及具体的命令是什么,同时他又描述了一些自己的做过的尝试,并且排除了自己认为可能的出现问题的原因,最后还上传了开发者想要的测试文件。以下是开发者的第二次回复,已经涉及到了关于这个问题的一些具体解释。 Ah, I thought you somehow suggested that transcripts were duplicated (the fix mentioned in the release notes for v1.3.1 was about that), but now I see that your problem is that genes are duplicated in the "gene abundance" output file. That's actually not quite true; each line in that output file is rather about estimating abundance for"gene"regions (loci), formed by overlapping transcripts, and in some cases such gene regions are not uniquely identified by their ID/name, the coordinates (genomic location) of each such"gene region" (locus) sometimes make a very important distinction. Look at the transcripts for that gene (AT1G58520) in the annotation file: there are 7 transcripts there, but one of them (AT1G58520.3) is actually not overlapping any of the others, so StringTie treats it as a separate locus ("gene"), and thus it creates a separate entry in the gene abundance file for that particular locus (AT1G58520|Chr1(+)21729913-21731344) which is different from the other, "main" locus which has all the other overlapping transcripts: AT1G58520|Chr1(+)21732566-21738808 StringTie cannot "trust" the reference annotation, as sometimes the gene ID can be just a duplicated string providing no true locus identity.. So you can think of it as StringTie splitting that "gene" into two non-overlapping gene regions and assessing the expression for each gene region independently. Again, the identity of a "gene" (actually "locus") in that file is given by gene ID and the genomic location of the underlying cluster of transcripts which define that locus. As you can see, that single transcript gene region (locus Chr1(+):21729913-21731344) has zero coverage so I don't know if that gene region is even real (or perhaps just an annotation artifact). If you really do not care about these situations (where gene definitions are not quite consistent with the transcripts so StringTie separates them like this), you could just add up the coverage values for all these lines with the same gene ID in the gene abundance file and call that the "total" abundance of that "gene" -- but this could be really misleading if the gene ID is not uniquely identifying a locus, i.e. if it's duplicated in other places on the genome (perhaps even on another chromosome). 这一段的作者的回答信息量比较大,和我之前写到的实际类似,stringtie 本身给你的所谓基因定量结果,看的并不是基因(名),而是基因的位置,如果你的两坨转录本没有交集,就会被自动划分为两个位置进行定量。作者特别用拟南芥的注释文件来举了个例子,并且说道我的软件可不会真的相信注释文,因为有的时候基因注释文件中的 id 会出现重复的情况,所以你可以理解为 stringtie 把一些转录本没有重叠的基因分成了两个部分来独立定量。我们在意的是 locus。最后作者也给了一个建议,如果你不在乎,可以把这些基因手动的合并一下,但是如果一个基因竟然出现在基因组的两个位置,这么做似乎也没什么意义。 然后,提问者并没有就此停下,他又提到了一个新的疑问,也就是我在上文有提到的,不同的样本重复情况不尽相同。这又是什么原因。 Ok, that makes a lot of sense, I like that stringtie identifies genes this way so that it does not combine only on a gene ID. What I am seeing though is that stringtie will "duplicate" some genes in only some of the output files. It would make sense if it did it all the time, but I only see it happening in some of them. I guess what I am concerned about is that stringtie is Identifying different amounts of genes each time. So far for arabidopsis I have seen it identify either 37363 or 37364 genes. From your last response, it seems that it should be identifying scenarios like the AT1G58520 and the AT1G58520.3 every single time it is run, not just occasionally. I was looking back at my data, and it appears that yes, this is true for AT1G58520, but it is not for ATCG09900. At the bottom of this response I have a grep of my gene abundance files for gene ATCG09900, and it appears that in some samples stringtie identifies 2 copies, but sometimes only 1. I looked at the reference.gtf, and it looks like it should have 2 copies every time like AT1G58520 because even though it has the same gene ID, it has different genomic location. Your response above makes a lot of sense to me, stringtie should require both gene ID and genomic location to avoid bad reference.gtf files. My problem now is that I don't understand why in the case of ATCG09900 it is not being consistent across samples? I would think that the part of stringtie that identifies the gene from the reference.gtf would be independent of the sample. Here is the reference.gtf for gene ATCG09900, I would expect that stringtie would always identify it as 2 genes from your last response: Here is a section of a grep of my output gene abundance files that shows that ATCG09900 is sometimes represented by 2 copies, and sometimes only 1. It seems that its representation as 2 copies is independent of if it is actually expressed or not. 这里提问者再一次详细的贴出了自己的实际分析结果,也非常清楚的说明白了自己困惑。于是,开发者又进行了进一步的回答。 Good question -- apologies for leaving out an important piece of information in my previous explanation: the fact that overlaps with read alignments from the BAM file are also taken into account when determining a "gene region" (locus), so they can act as a "glue", or "bridge" which could put together "gene regions" otherwise separated when looking only at overlaps between reference transcripts. So it is likely that in some samples the two gene regions of ATCG09900 get "bridged" by read alignments overlapping both those regions.. Thus only one gene region is reported for such samples. It is all related to the concept of "bundle" as used by StringTie. Read alignments and reference transcripts are binned together in a "bundle" defined as a transitive closure of the exon overlap relationship between these objects (read alignments or reference transcripts (guides)). StringTie analyses a "bundle" and unless "weak spots" are found (spurious alignments) to break the bundle into multiple regions, the "bundle" will end up as a "gene region" (locus) reported in the output. This "clustering by overlap" approach can also have the downside of merging multiple otherwise clearly distinct gene regions together (i.e. with different reference gene IDs), when alignment artifacts and/or transcriptional noise artificially "bridge" over intergenic regions, in some rare cases (especially when the actual genes are very close to each other). 开发者先感谢对方提出了这样一个好问题,他解释道自己有一个重要的统计细节没有讲,就是当决定基因范围时,stringtie 还会考虑实际的数据比对情况,reads 可以作为胶水将两个分离的基因区域连接起来,如果有这样的情况,这个基因也不会被分开。当然,最后作者也说了这样处理数据的一些缺陷。 这次,提问者的困惑被解决了,他对自己的问题进行了一个自我总结作为最后的回复,也作为对开发者回答的响应。内容如下: Ahh, very good. So the samples where there is only 1 gene region means that there was either a bridge between the two transcripts, or there was no spurious alignment to break apart the bundle (in my case, this often meant no alignment). With that info, I think I am able to answer my last question on my own. Above, some of the samples has genes that had fpkm of 0, but they still split, while others had fpkm of 0 and did not split. For example: It would appear that SRX2248582 and SRX2248583 should both fail to break the bundle since no alignment has happened in either that would cause the bundle to break. This is the case in SRX2248583 but not in SRX2248582. I went and looked at the sam file for these, and found that SRX2248582 had a read in that region, while SRX2248583 did not (I also checked the region 100 before, but none overlapped with the region so for brevity I will omit them): I suppose that this spurious alignment caused the split, but was not counted towards the fpkm for some other reason. Thank you for all of your help and quick response times, I appreciate it a lot. 在最后的总结中,作者又提到了一个观察到的现象:一个基因在不同的样本中定量的结果都为 0 ,但是有一些样本中这个基因被拆分为二,另外一些样本则没有。他认为这可能就是开发者提到的是否存在一些 reads 比对的问题,于是他又检查了不同样本中这个基因所在位置区间的比对情况,果然和开发者提到的内容相符。 写到这里,忍不住在 GitHub 上评论了一下,虽然 issue 已经关闭了。 这次 debug 我最先猜测到的还是问题的表象,然后通过一些后续的分析找到了可能的问题。在看到上面这个完整的 issue 后,我深切地感受到了什么是一个优秀的提问,他可以指引被提问对象说出关键的信息,并且循序渐进的做出需要的补充内容。如果没有提问者详细的描述和认真的思考,可能这个问题不知道还要经历多少个来回才能被讨论清楚。 经历了这么一番波折,我对这个工具的喜欢竟然增加了一点,接下来就是再仔细看看文章和代码。也许,这就是为什么人总是会对伤害过自己的人和事念念不完。Bioinformatics was like a box of chocolates. You never konw what you're gonna get ——阿飞正传 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名 - 非商业性使用 - 禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。
铺垫
尝试
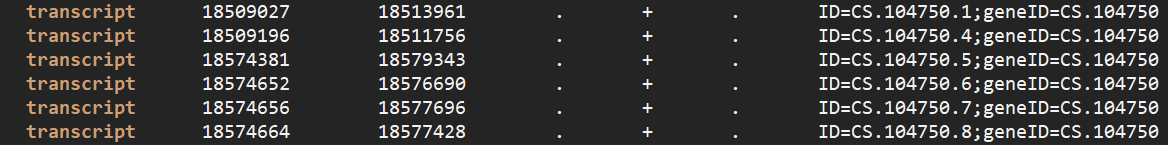
CS.104750 在最后的表达定量中出现了两次,且一个基因给出的位置坐标是在转录本 1 和 4 上(18509027-18513961),另一个基因位置是在 4-8 上(18574381-18579343)。如果是通过 ID 来拆分转录本应该是转录本 1 独立为一个基因,其它转录本独立为一个基因。这里似乎是按照位置进行了区分,因为转录本 1、4 和另外 5-8 之间没有位置上的重叠,所有被分为了两个基因。在检查其它几个重复出现的基因,都符合我这个猜想。惊喜
stringtie gene abundance duplicate 。用这几个关键词,Google 第二个结果 就是一个和我完全一样的困惑(似乎也只有这一个内容和我的困惑相关),而且也是在 GitHub 上提出的问题。
stringtie -v -p 1 -e -o test.gtf -G TAIR10_Araport11.gtf -A test.ga -l test OutputFromHisat-Samtools_sort-Samtoools_Index.bam
Araport11_GFF3_genes_transposons.201606.gff.gz
gffread -E Araport11_GFF3_genes_transposons.201606.gff -T -o- > TAIR10_Araport11.gtf
ATCG09900
AT1G79790
AT1G58520
SRX2248274/SRX2248274_vs_TAIR10_Araport11.ga
SRX2248275/SRX2248275_vs_TAIR10_Araport11.ga
AT1G58520
SRX2248276/SRX2248276_vs_TAIR10_Araport11.ga
AT1G58520
SRX2248277/SRX2248277_vs_TAIR10_Araport11.ga
AT1G58520
SRX2248278/SRX2248278_vs_TAIR10_Araport11.ga
AT1G58520
SRX2248279/SRX2248279_vs_TAIR10_Araport11.ga
AT1G58520
ATCG09900
SRX2248280/SRX2248280_vs_TAIR10_Araport11.ga
AT1G58520
SRX2248281/SRX2248281_vs_TAIR10_Araport11.ga
AT1G58520
SRX2248282/SRX2248282_vs_TAIR10_Araport11.ga
AT1G58520
SRX2248283/SRX2248283_vs_TAIR10_Araport11.ga
AT1G58520
ATCG09900
SRX2248284/SRX2248284_vs_TAIR10_Araport11.ga
AT1G58520
ATCG09900
SRX2248285/SRX2248285_vs_TAIR10_Araport11.ga
AT1G58520
SRX2248286/SRX2248286_vs_TAIR10_Araport11.ga
AT1G58520
SRX2248287/SRX2248287_vs_TAIR10_Araport11.ga
AT1G58520
stringtie -v -p 1 -e -o test1.gtf -G TAIR10_Araport11.gtf -A test1.ga -l catz bundle.bam
awk '{print $1}' test1.ga|sort |uniq -d
# AT1G58520
ChrC Araport11 exon 7967 7996 241.00 - . transcript_id "ATCG09900.1"; gene_id "ATCG09900"; gene_name "ATCG09900";
ChrC Araport11 exon 8176 8207 241.00 - . transcript_id "ATCG09900.2"; gene_id "ATCG09900"; gene_name "ATCG09900";
SRX2248582/SRX2248582_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 7996 0.000000 0.000000 0.000000
ATCG09900 ATCG09900 ChrC - 8176 8207 0.0 0.0 0.0
SRX2248583/SRX2248583_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 8207 0.0 0.0 0.0
SRX2248584/SRX2248584_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 7996 1.533333 0.790024 1.578678
ATCG09900 ATCG09900 ChrC - 8176 8207 0.0 0.0 0.0
SRX2248585/SRX2248585_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 8207 0.0 0.0 0.0
SRX2267752/SRX2267752_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 8207 0.0 0.0 0.0
SRX2267753/SRX2267753_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 8207 0.0 0.0 0.0
SRX2267754/SRX2267754_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 8207 0.0 0.0 0.0
SRX2267755/SRX2267755_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 7996 0.333333 0.276271 0.562470
ATCG09900 ATCG09900 ChrC - 8176 8207 0.875000 0.725212 1.476486
SRX2267756/SRX2267756_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 7996 0.333333 0.177870 0.363462
ATCG09900 ATCG09900 ChrC - 8176 8207 0.281250 0.150078 0.306671
SRX2267757/SRX2267757_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 8207 0.0 0.0 0.0
SRX2267758/SRX2267758_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 8207 0.0 0.0 0.0
SRX2248582/SRX2248582_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 7996 0.000000 0.000000 0.000000
ATCG09900 ATCG09900 ChrC - 8176 8207 0.0 0.0 0.0
SRX2248583/SRX2248583_vs_TAIR10_Araport11.ga
ATCG09900 ATCG09900 ChrC - 7967 8207 0.0 0.0 0.0
$ grep ChrC *.sam| awk -F "\t" '{ if(($4 >= 7967 && $4 <= 8207)) { print } }'
SRR4426896.15866700 16 ChrC 8000 60 9M1I90M * 0 0 ATATATATATTTTTTTTTCATTTTCTATATTTTTTTCTATATTTTATTATATTATTATATATATATATATTCTTTTTGATTATTTGATTATATAAATATA CEEDEEEDDDDDDFFFDDDHHGGHHJJJJIGIJIIJIJJJJJIJIHIIGIGGIGJJJJIJJJJJJJJJJJJJJJJJJJJJJJIJJJJHHHHHFFFFFCCC AS:i:-8 ZS:i:-10 XN:i:0 XM:i:0 XO:i:1 XG:i:1 NM:i:1 MD:Z:99 YT:Z:UU NH:i:1
$ cd ../SRX2248583
$ grep ChrC *.sam| awk -F "\t" '{ if(($4 >= 7967 && $4 <= 8207)) { print } }'
反思
· 分享链接 https://kaopubear.top/blog/2019-05-07-stringtiegenetpm/