生物统计学与R极简手册
写在前面
入门生物信息或者进行生物相关研究,所有人都绕不开统计的基础知识和计算实现方式。在担任中科院生物统计学课程助教的过程中,我发现大部分同学的首要困惑在于理不清相关概念,其次才是不知道该如何用R语言来进行最基本的计算。本合集共分为8小节,将简要介绍生物统计学相关基础知识以及如何使用R语言进行最基本的计算和分析。
需要说明的是,文中个别描述严格来讲并不准确但希望有助于理解,涉及到R语言的部分则展示了若干函数最基本用法,希望不给阅读和学习增加负担。另外,这份资料主要面向生物统计学和R语言基础薄弱的人群,勉强可以称之为极简手册 ,详细的学习还需要阅读相关教材资料。
生物统计学与R极简手册写在前面描述性统计量数据的集中趋势数据的变异性(离散性)计算描述性统计量形象化展示概率相关内容几个概念离散概率分布常见的离散概率分布连续概率分布估计相关性分析正态分布评估相关性分析相关性可视化展示Pearson 相关性检验单双样本均值分析假设检验一般步骤单样本t-test单样本Wilcoxon 符号秩检验独立双样本t-test独立双样本Wilcoxon test非独立双样本t-test非独立双样本Wilcoxon test多样本均值分析one-way ANOVAtwo-way ANOVA比例分析单比例检验双比例检验卡方分布常用高阶分析方法回归分析聚类分析k-meansHierarchical Clustering主成分分析
描述性统计量
为了解决某个问题,我们通常会观察一组和该问题相关的样本,利用总体中的部分样本来推断总体的情况进而得到相关结论。在通过样本推断总体前,首先需用对已有样本数据进行简单的评估和描述,针对这一需求也就引出了描述统计量这一概念。进行描述性统计时,我们最关注数据两个层面的问题:数据的集中趋势和变异分散性。
数据的集中趋势
面对少则几十多则上千个数字,第一步通常是观察平均水平。下面介绍三个计算数据平均水平的概念:分别是均值(mean)、中位数(median)和众数(mode)。
均值:所有观察值的和除以观察的个数。算数平均是最自然和常用的测度,其问题在于对异常值(outliers)非常敏感。有极端值存在时,均值不能代表样本的绝大多数情况。
中位数:所谓中位数,是指所有样本观测值由小到大排序,位于中间的一个(样本数为奇数)或者两个数据的平均值(样本数为偶数)。
当数据分布对称时,中位数近似等于算数平均数;当数据正倾斜时(图像向右倾斜),中位数小于算数平均数;当数据负倾斜时(图像向左倾斜),中位数大于算数平均数。因此,我们可以通过比较样本的均值和中位数对数据的分布对称性进行初判断。
众数:在样本所有观测值中,出现频率最大(出现次数最多)的数值称为众数。这里需要说明,当数据量很大而且数值不会多次重复出现时众数并不能带来太多信息。比如当计算上万个基因的表达量后,得到的众数最可能是0,因为每个基因的表达值或多或少都有一些不同,这时候出现最多的就是那些没有检测到表达基因的0了。但是在遇到类别数据而非数值型数据时众数有很大用处,或者说众数是唯一可以用于类别数据的平均数。
在R中,均值和中位数可以通过mean()和median()进行计算,而众数可以通过modeest包mfv()函数得到。
数据的变异性(离散性)
平均数显然不能说明一切问题,在说明样本数据时我们还必须考虑数据是不是过于分散。例如在篮球队员投篮平均得分相同的情况下,更重要的是知道他们谁发挥更加稳定。
极差(range)指一个样本中最大值和最小值之间的差值。在统计学中也称为全距,它能够指出数据的“宽度”(范围)。但它和均值一样易受极端值影响,而且也会受样本量明显影响。
针对极差的缺点,统计学又引入分位数(quantiles)概念,通俗讲是把数据的“宽度”细分后再去进行比较从而更好地描述数据的分布形态。分位数用三个点将从小到大排列好的数据分为四个相等部分,而这三个点也就是我们常说的四分位数,分别叫做下四分位数,中位数和上四分位数。当然,除了四分位也可以计算十分位或者百分位。
分位数的引进能够说明数值的位置,但无法说明某数值在该位置出现的概率。为了说明数据的稳定程度,我们可以考虑计算每个数据值到平均数的距离(此处可以脑补一个高瘦形的数据曲线和矮胖形的数据曲线),但样本中所有观测值与均值偏差的和永远是0。为了解决这种正负距离相互抵消的问题,统计学又引入方差(variance)和标准差(standard deviation)概念。
所谓方差指数值与均值距离平方数的平均数,而标准差则是方差的平方根。标准差体现了数据的变异度,标准差越小,数值和均值越近。通常均值用表示,而标准差用表示。
在R中,可以通过quantile()计算分位数,通过var()来计算方差,通过sd()来计算标准差。
有了标准差的概念,随之而来的问题是当两个样本标准差相同但是均值相差很大时该如何做出区分。于是,统计学引入了变异系数(coefficient of variation, CV) 概念,变异系数是指样本标准差除以均值再乘100%。变异系数不会受数据尺度的影响,因此常用来进行不同样本之间变异性的比较。
在实际的数据分析中,如果要比较不同数据集(均值和标准差都不同)之间的数值,通常会引入z score的概念,z score 的计算方法是用某一数值减去均值在除以标准差。通过对原始数据进行z变换,我们将不同数据集转化为一个新的均值为0,标准差为1的分布。
计算描述性统计量
在R中,使用summary()函数会得到一个data frame 的很多 描述性统计量。当数据某一列是数值型变量时,可以得到该列数据的均值、极值、方差和分位数。
下面我们使用R中内置的数据Edgar Anderson's Iris Data进行一些简单展示。
xxxxxxxxxxsummary(iris)#查看常用的描述统计量xxxxxxxxxxSepal.Length Sepal.Width Petal.Length Petal.Width SpeciesMin. :4.300 Min. :2.000 Min. :1.000 Min. :0.100 setosa :501st Qu.:5.100 1st Qu.:2.800 1st Qu.:1.600 1st Qu.:0.300 versicolor:50Median :5.800 Median :3.000 Median :4.350 Median :1.300 virginica :50Mean :5.843 Mean :3.057 Mean :3.758 Mean :1.1993rd Qu.:6.400 3rd Qu.:3.300 3rd Qu.:5.100 3rd Qu.:1.800Max. :7.900 Max. :4.400 Max. :6.900 Max. :2.500
形象化展示
所谓形象化展示就是用图来展示数据结果,比较常见的方法有条形图,箱线图,直方图等
xxxxxxxxxxboxplot(iris$Sepal.Length)# 使用箱线图展示某一列数据的分布情况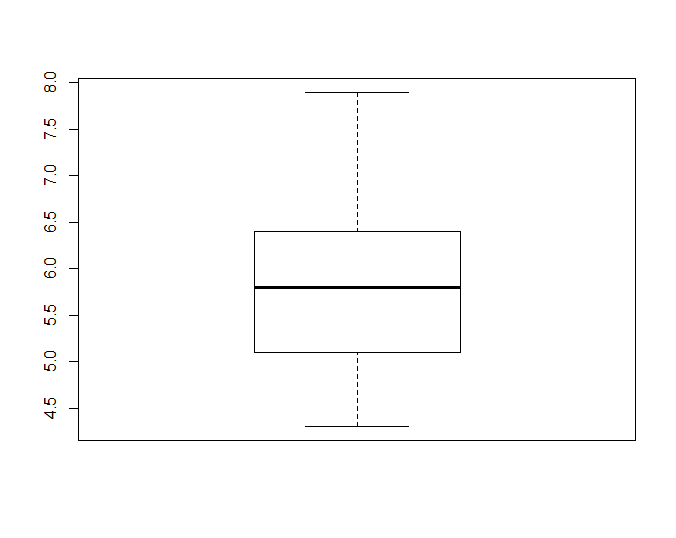
xxxxxxxxxxhist(iris$Sepal.Length)# 使用直方图展示某一列数据的分布情况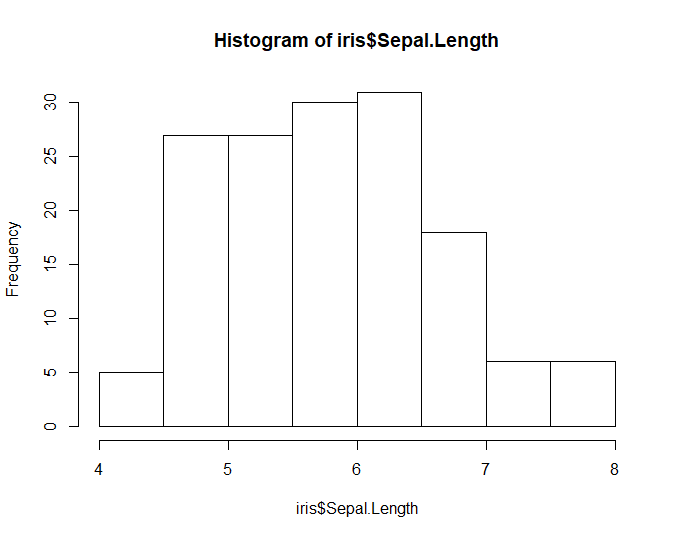
xxxxxxxxxxplot(ecdf(iris$Sepal.Length))# 绘制简单的累积分布函数图展示某一列数据分布情况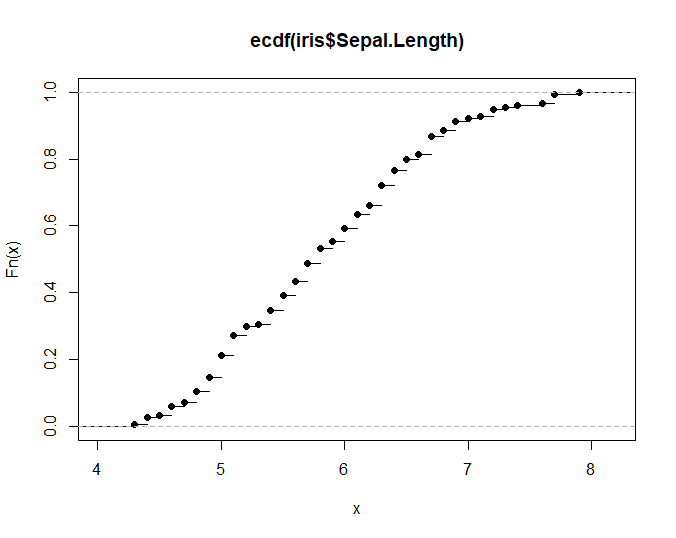
概率相关内容
统计学中大量内容源于概率,学习统计学也就必须要了解一些概率中的基本概念,其中尤为重要的是条件概率,以及延伸出的贝叶斯定理(也许是最牛也是最难充分掌握的内容)。
几个概念
样本空间(sample space)是所有可能结果的一个集合,事件(event)则是样本空间中所有感兴趣结果的一个子集。某事件的概率(probability)是该事件在无限次试验次数中的相对频率。
事件的交集并集和补集,概率的加法法则和乘法法则在本章中不再做介绍。
条件概率(Conditional Probability)用来描述与其他事件的发生相关的某事件的概率,通常被描述成“在A事件下发生事件B的概率”。在书写时用|表示,例如P(A|B)是在B发生的情况下A发生的概率。计算方式为AB同时发生的次数除以所有B发生的次数。
条件概率计算方法:
可推出: =>
全概率公式: (贝叶斯定理分母部分)
贝叶斯定理:
得到的贝叶斯定理可以帮助我们计算逆条件概率。
在实际的生物学数据处理的过程中,我们还会接触到灵敏度(sensitivity)、特异度(specificity)、假阳性(false negative)和假阴性(false positive) 几个概念。在疾病相关研究中,对于某一个症状,灵敏度指发病后出现症状的概率,特异度是不发病时不出现症状的概率。假阳性是指实验结果阳性但是实际为阴性,假阴性是指实验结果为阴性但是实际为阳性。基于灵敏度和特异度可以使用ROC曲线,通常来说在两个检验中,曲线下面积大的较好。
概率能够告诉我们事件发生的可能性,但如果想要利用概率预测未来的结果并且评估预测的确定性就需要引入概率分布。
离散概率分布
随机变量: 样本空间中,对不同事件指定有相应概率的数值函数。
随机变量是可以等于一系列数值的变量,这些值都和一个特定概率关联。它的写法是,表示随机变量X取特定值为x时的概率。随机变量包括离散和连续两种形式,所谓离散指变量只能取一些确定值。连续指的是有无限多种可能取值。
概率分布也叫概率质量分布,。在描述统计量中,样本的频数分布描述每个取值及对应发生次数,如果样本总数除以对应发生次数,得到的频率分布就类似于这里的概率分布。后面会提到的“拟合优度检验”就是比较有限样本频率分布和概率分布的差异。
如果将随机变量和样本对应起来理解,样本中均值的概念在总体(随机变量)中称为期望,也叫作总体均值,表示为(和均值一致)或者。计算方式是将每个可能值和概率相乘再把所有乘积相加。和均值类似,这里的期望也无法描述相关数值分散程度。
同样,在随机变量中也有类似于样本方差的概念,称为总体方差(随机变量方差),用来表示分散程度。其计算公式为 。而概率分布的标准差同样是方差的平方根。
计算时,首先计算每个数值x的,然后再将结果乘以概率,最后把所有结果相加。
在数据集中,方差和标准差表示的是数据和均值的距离,在概率分布中表示特定数值概率的分散情况。方差越小,结果越接近期望。
累加分布函数(cumulative-distribution function, cdf): 随机变量X,对于X的任一指定值x,概率值。即随机变量取值不大于指定值的概率。记作
常见的离散概率分布
几何分布:进行一组相互独立实验,每次实验有成功失败两种可能且每次试验概率相等,想知道第一次成功需要进行的试验次数。
期望;方差
二项分布:进行一组相互独立试验,每次实验有成功失败两种可能,每次试验概率相等且试验次数有限,想知道在有限次试验中成功的次数。 期望;方差
二项分布和几何分布差别在于,前者试验次数固定求成功概率,后者求第一次成功前试验次数。
泊松分布:常与稀有事件相关,单独事件在给定区间(区间可以是时间或者空间)内随机独立发生,该区间内的事件平均发生次数已知且为有限值。这个值用表示。给定区间内发生r次事件的概率计算公式: 期望是给定区间内能够期望的事件发生次数λ,方差也是λ。如果一个离散随机变量的一批数据计算后方差和均值近似相等,则可以推测样本符合泊松分布。
当二项分布的p很小且n非常大时,,方差和期望近似相等,可以用泊松分布来近似二项分布,从而使计算简化。通常,n大于50且p<0.1时为典型的近似情况。
连续概率分布
当数据连续分布,人们关心的是取得某一个特定范围的概率。
概率密度函数(probability density function, pdf):本质是一个函数,用这个函数可以求出在一个范围内某连续变量的概率,同时该函数可以指出该概率分布的形状。换句话讲,任意a,b两点之间及函数对应曲线下组成的面积等于随机变量X落在ab间的概率。曲线下面积总和是1。
概率密度可以指出各种范围内的概率大小,用面积来表示。概率密度只是表示概率的一种方法而非概率本身。
累加分布函数a点上的值等于随机变量X取值的概率,也是概率密度函数a左边曲线下的面积。
正态分布是连续数据的一种理想状态。正态分布的概率密度函数是一条对称的钟形曲线,均值具有最大的概率密度,偏离均值概率密度逐渐变小。参数是均值,也是曲线的中央位置,表述曲线的“胖瘦”。
连续随机变量X符合均值为,标准差为的正态分布时写作.
线性变换:
当X和Y相互独立(彼此之间没有影响)时: 期望;方差
当X和Y并不彼此独立时,使用协方差(Covariance)来描述两个随机变量间的关系,记做Cov(X,Y)
二项分布正态近似:通常情况,当二项分布满足 (也有建议)时,可以用正态分布代替二项分布。其中,。另外,当泊松分布的λ>15时,也可以用正态分布进行近似。
估计
在通常的试验中我们获得的信息总是来自样本,想要知道总体的参数,只能通过已有样本参数进行估计。
样本均值是总体均值的点估计,通常样本均值用表示,总体均值用表示。
在估计总体方差时,计算公式为
用样本方差估计总体方差会使得估计结果偏低,样本越小两个方差的差别可能就越大。在这里,估计总体方差的公式中除的是,能够更接近总体方差。另外,总体方差点估计公式通常记做,写作:
用样本均值估计总体均值时也会产生误差,均值的标准误差是,估计量是。标准误差表示了样本均值的分散情况,从公式中我们可以看出,n越大,用样本均值估计总体均值越准确。当样本足够大(大于30),即便总体不符合正态分布,但从中取出的样本均值分布仍然近似于正态分布(中心极限定理)。
除了对总体进行点估计以外,我们往往还会对总体进行区间估计,即对通过点估计得到的结果加减一定范围的误差。
相关性分析
本节提到的相关性分析和后面会提到的t-test, ANOVA 以及回归分析等被称为参数检验,这些检验在进行时我们常默认数据符合一定前提条件,如符合正态分布和方差相等等。当样本数量大于30时,根据中心极限定理,我们通常认为数据符合正态分布;在进行t-test 和ANOVA 分析时,还需要满足样本方差相等。
在进行各种检验之前需要初步检验数据是否符合某种检验的前提条件,如果不符合则应该考虑使用非参数检验或其他方法。
正态分布评估
在评估数据集是否符合正态分布时通常会采用Shapiro-Wilk’s test和图示(Q-Q plot)结合的方法。使用Q-Q plot(quantile-quantile plot)的结果较直观,使用Shapiro-Wilk’s test显著性检验的方法更准确(相对而言)。
Shapiro-Wilk’s test 结果受样本量的影响非常大,当样本量很大时即便数据符合正态分布也容易出现p值很小进而拒绝原假设的情况(该检验原假设是样本来自于正态分布)。样本量很小时即便真实数据不s是来自正态分布,也可能接受原假设。
这里试举一例
xxxxxxxxxx# 分别随机生成两组二项分布和指数分布随机数set.seed(90)x <- rbinom(15,8,0.7)y <- rexp(15,0.5)shapiro.test(x)shapiro.test(y)# Shapiro-Wilk normality test## data: x# W = 0.95996, p-value = 0.6917### Shapiro-Wilk normality test## data: y# W = 0.96168, p-value = 0.7216可以发现,即便我们生成的两个样本都不是正态分布,但是检验的结果仍然没有拒绝原假设(没有拒绝不等于接受原假设)。好在R中该函数限制检测的样本个数是3到5000。因此,同时结合图像来分析还是很必要的。
一般使用Q-Q plot来检验是否符合正态分布,R中默认的函数是qqnorm();ggplot2中可以使用函数qplot();qqpubr包是基于ggplot2二次开发的简易升级版,操作更加友好,可以使用函数ggqqplot()。
下面利用生成的数据绘图。
xxxxxxxxxx# 生成符合正态分布的一组数据并绘图z <- rnorm(50)qqnorm(z)library(ggplot2)qplot(sample=z)library(ggpubr)ggqqplot(z)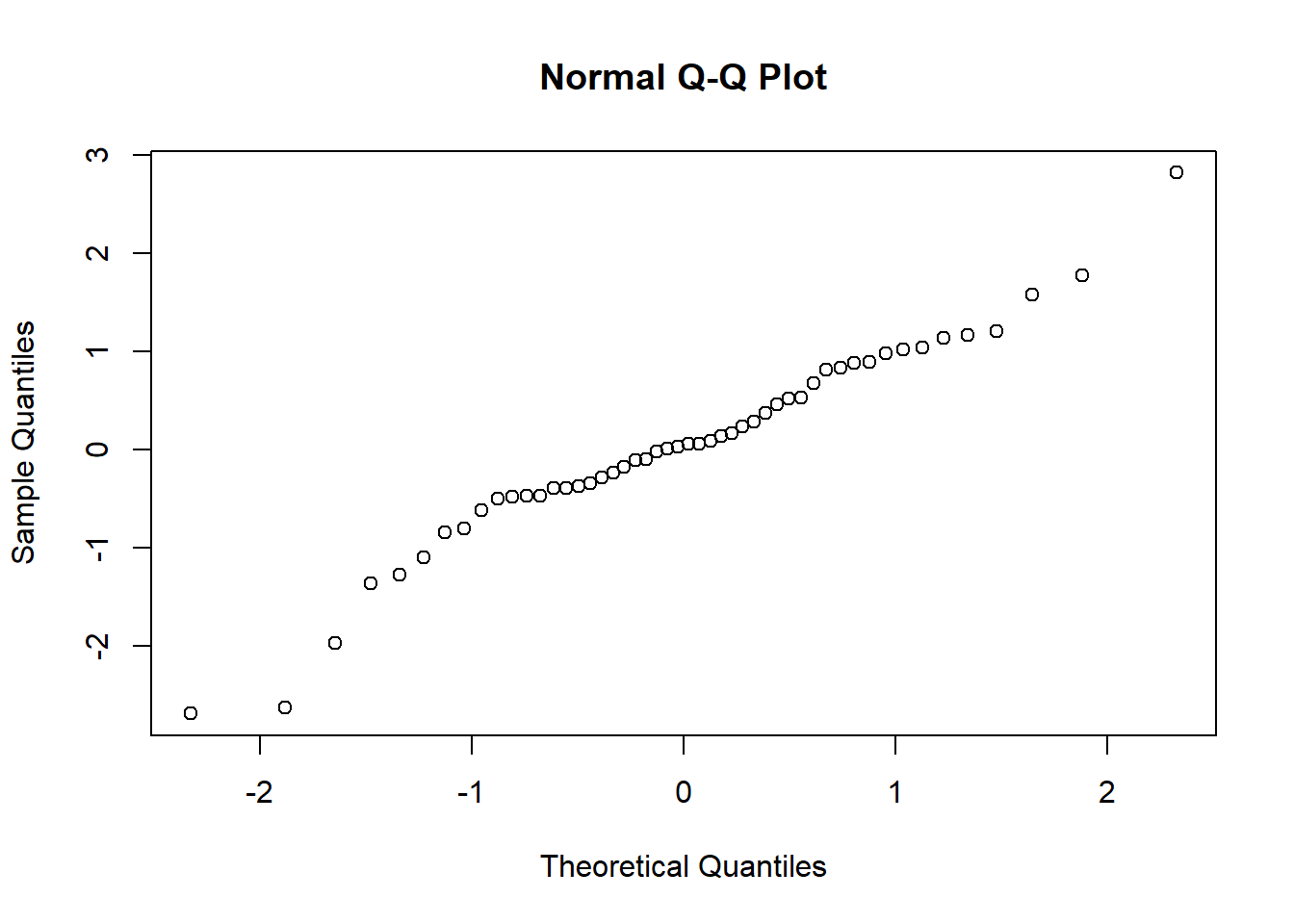
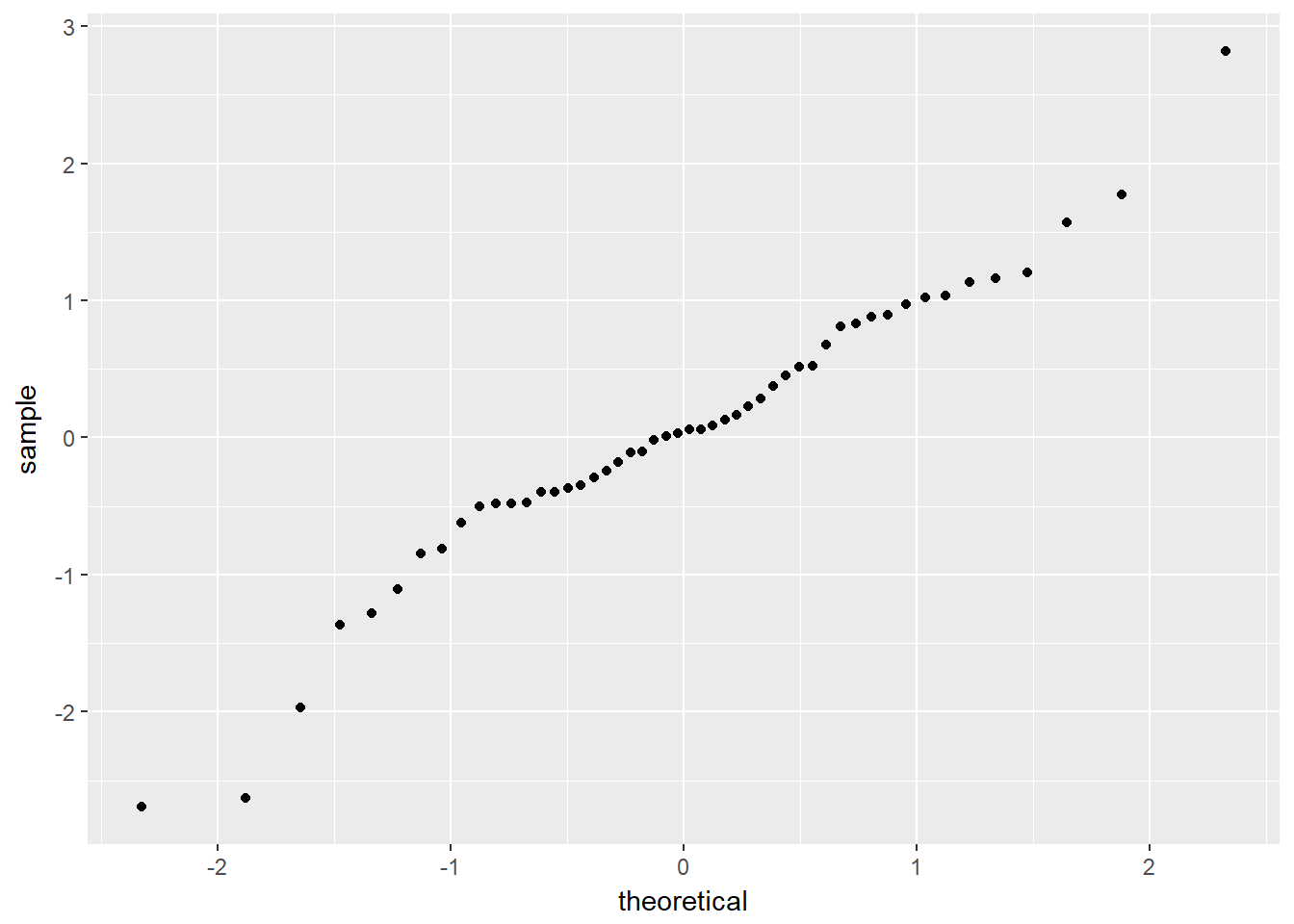
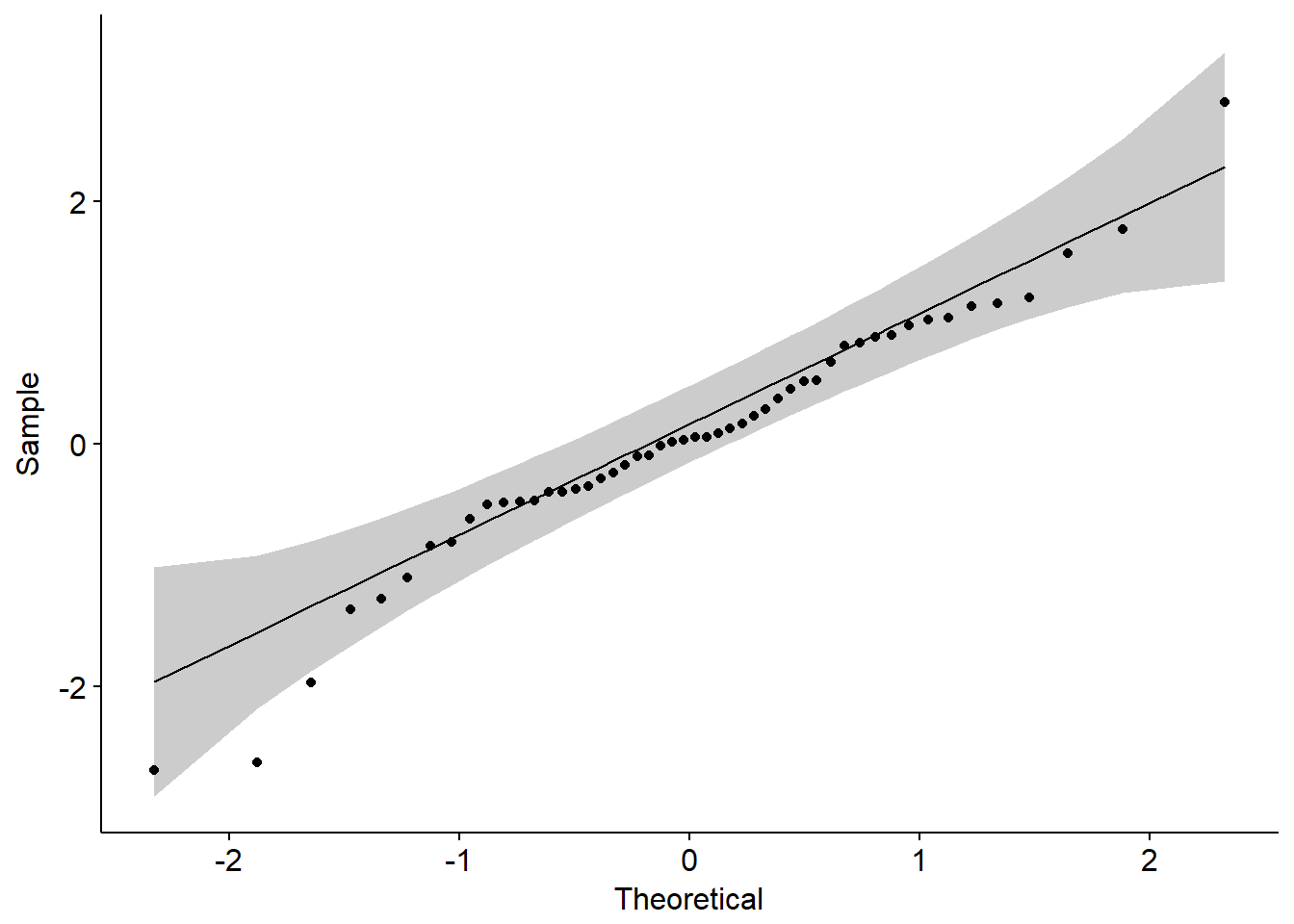
相关性分析
Pearson相关系数、Spearman相关系数、Kendall相关系数都可以用来表示变量之间的相关性,一般情况使用pearson相关系数更多,如果明确样本不符合正态分布可以使用kendall或者spearman相关系数。这三种相关系数都可以通过cor()函数来进行计算,下面通过R已有数据集cars,查看汽车车速和刹车距离之间的相关性。
pearson相关系数计算公式 其中m表示均值。
xxxxxxxxxxcor(cars, method = "pearson")相关性可视化展示
可以使用散点图进行两个变量之间的相关性展示。
xxxxxxxxxxplot(cars)ggplot(cars, aes(x=speed, y=dist))+ geom_point()ggscatter(cars,x="speed", y="dist",\add = "reg.line", conf.int = T,cor.coef = T)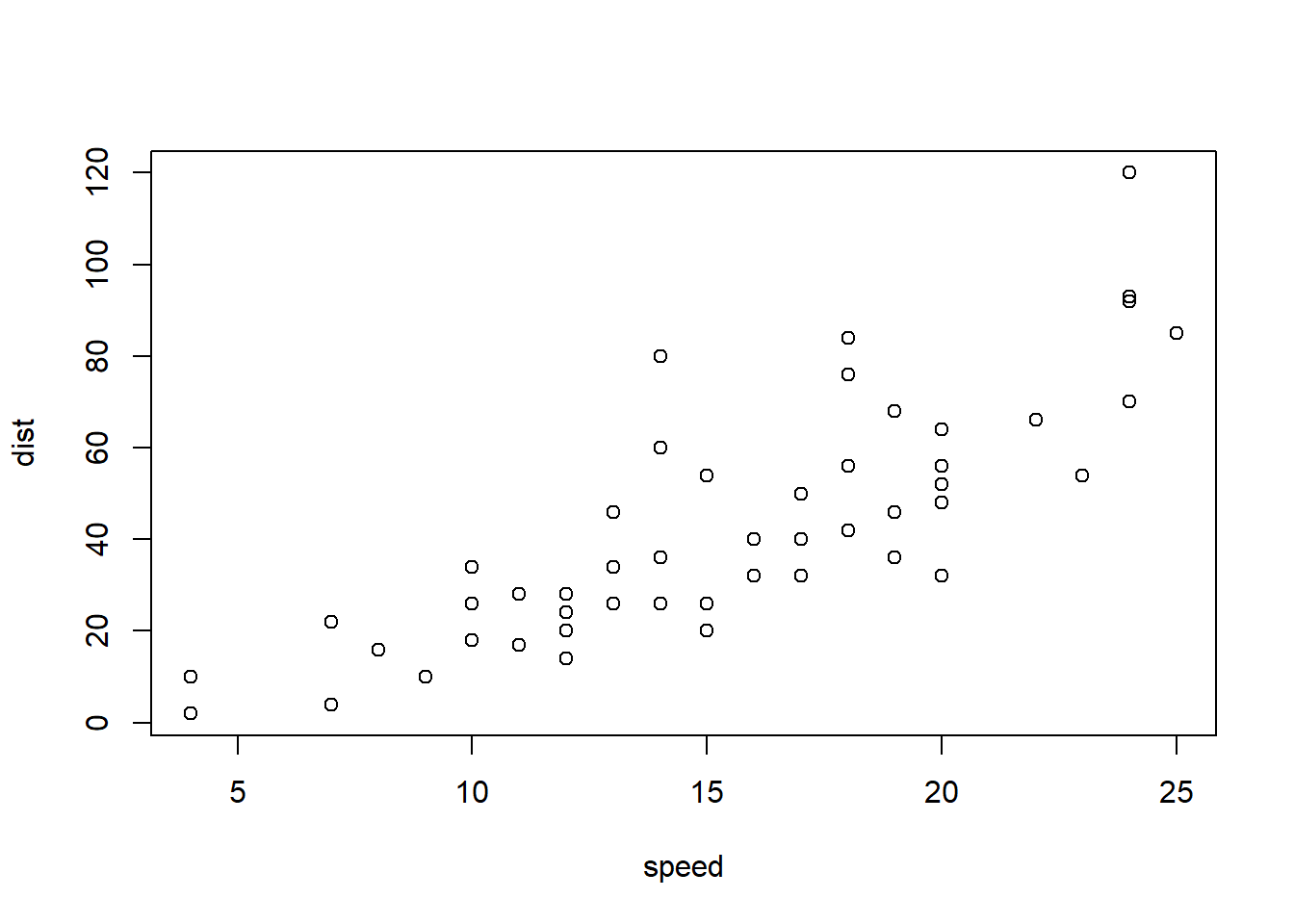
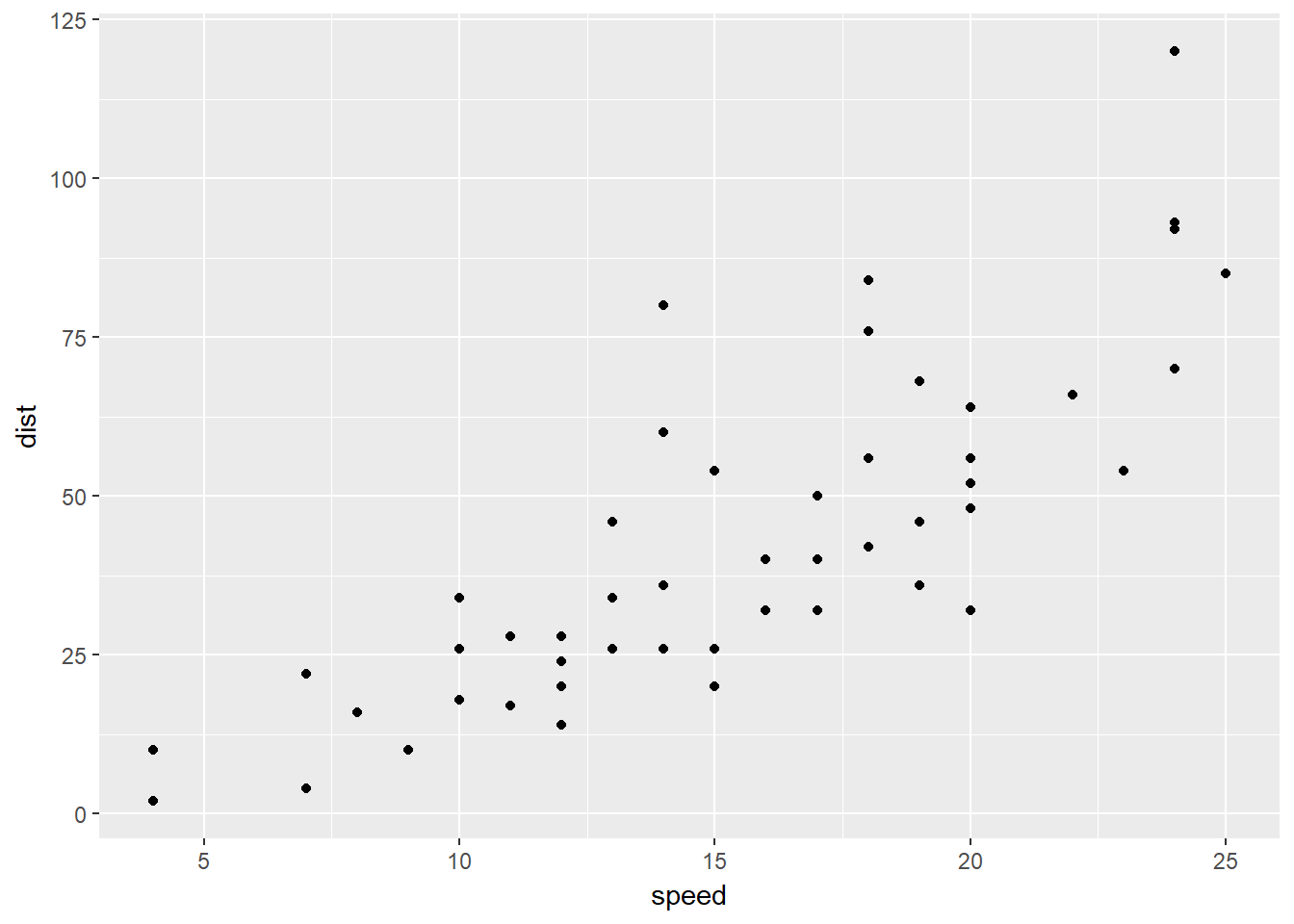
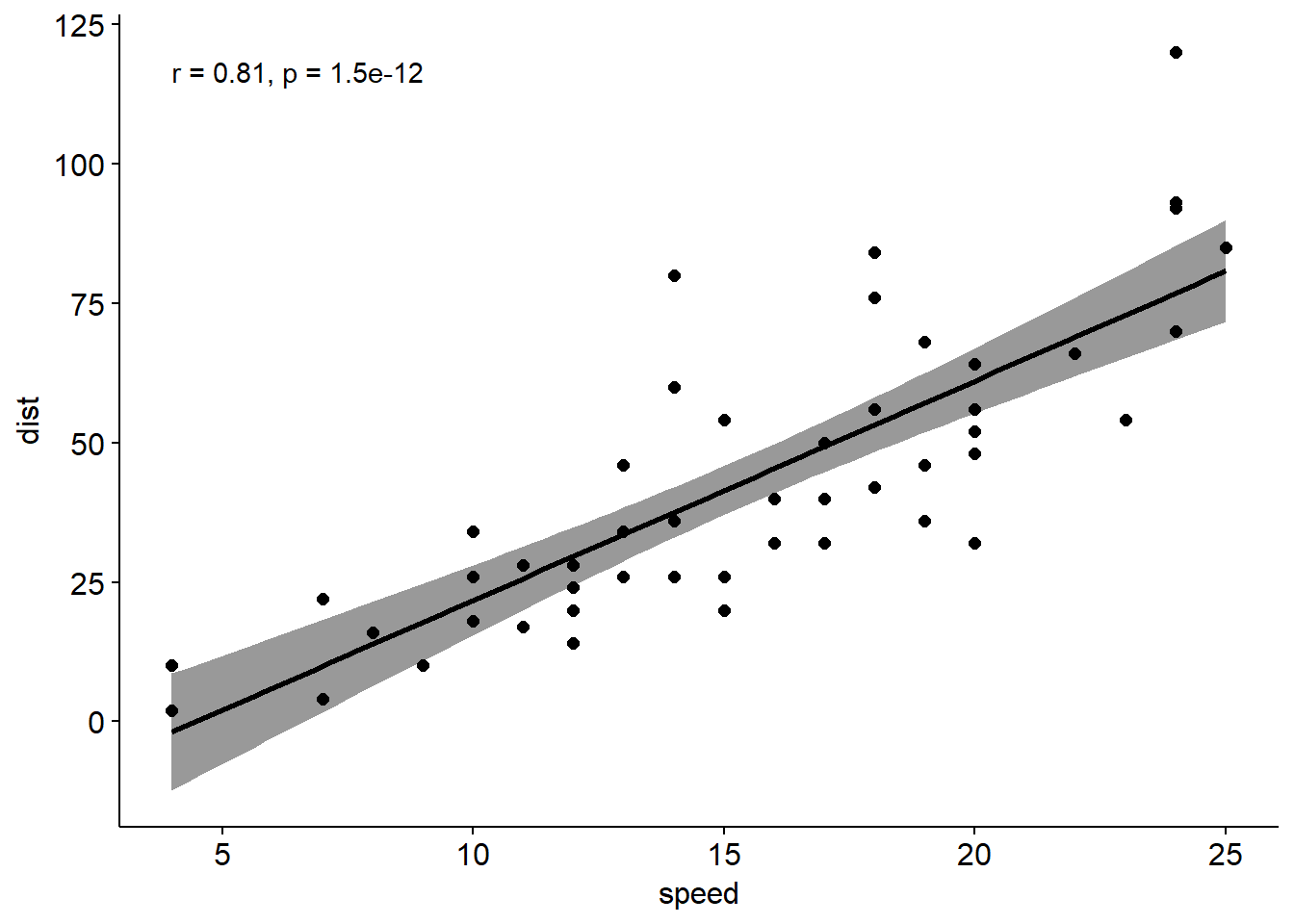
Pearson 相关性检验
前面我们只是计算了两个变量之间的相关性,还应该对相关进行显著性检验。原假设为变量之间没有相关性,使用函数为cor.test()
xxxxxxxxxxcor.test(cars$speed,cars$dist,\alternative = "two.side", method = "pearson")统计结果中,t表示t检验统计量,df表示自由度,pvalue是t检验的显著性水平,conf.int表示95%的置信区间,sample estimates 是相关系数。相关系数越接近-1表示负相关,接近1表示正相关。
单双样本均值分析
根据数据组数的不同,均值比较可以分为单样本、双样本和多样本。本节首先介绍单样本和双样本。
假设检验一般步骤
谈具体的假设检验之前首先介绍假设检验的一般步骤。
- 确定假设
原假设(null hypothesis, ) 指需要检验的假设,只要当我们有足够的证据时才能否定。在某种意义上与原假设相反的的假设被称为备择假设(alternative hypothesis,)。
假设检验是无法给出绝对证明的,我们只能在假定原假设为真的情况下通过假设检验来判断结果是否可信。如果结果极不可能发生,则拒绝原假设,进而接受备择假设。
根据接受假设和真实情况之间的关系,会有四种可能发生
| 情况 | 真实情况 | |
|---|---|---|
| 接受 | 为真且被接受 | 是真,被接受 |
| 拒绝 | 为真但是被拒绝 | 是真,被拒绝 |
其中为真但是被拒绝的错误概率称为I型错误,通常用表示,也称为显著性水平,为真但是接受的错误概率称为II型错误,常用表示。
检验的功效(power)=1-。虽然我们希望和都可以尽可能小,但这两者往往是矛盾的,通常我们会先固定在某个水平,再找合适的检验使尽可能小。
- 选择检验统计量
检验统计量(test statistic)是用于对假设进行检验的统计量,进行检验的过程建立在这个统计量之上。
- 决定拒绝域
拒绝域是可以不接受原假设的一组数值,其分界点被称为临界点。为了求出拒绝域首先要确定假设检验的显著性水平,也就是当样本结果的不可能发生程度多大时就可以拒绝原假设。
单尾检验指检验的拒绝域落在可能数据集的一侧(左侧或者右侧),双尾检验的拒绝域则分布在数据集的两侧,对于检验水平是的双尾检测而言,两侧分别是。如果备择假设的表示是“不等于”,则应该使用双尾检验。
- 求出p值
p值取决于检验统计量和拒绝域。表示得到更加极端结果的概率。
- 判断样本结果是否在拒绝域内
- 做出决策
单样本t-test
说完假设检验的基本流程之后,下面介绍和均值比较相关的假设检验,首先是单样本t-test。
在数据符合正态分布的前提下使用单样本t-test来比较一组样本的均值和已知(理论/总体)均值,所谓的已知均值可能来自于之前的实验数据或者理论值。根据研究问题(原假设)的不同又分为双尾(不等)和单尾检验(大于或者小于)。
统计量计算公式为
其中表示样本均值,n表示样本量,s是样本标准差(总体方差未知),表示理论值。通过统计量t和自由度,我们可以计算出对应的p值。
使用R进行单样本t-test,这里借用R自带数据集PlantGrowth的10个对照组数据。
xxxxxxxxxxweight <- PlantGrowth$weight[PlantGrowth$group=="ctrl"]shapiro.test(weight)summary(weight)# Shapiro-Wilk normality test##data: weight#W = 0.95668, p-value = 0.7475## Min. 1st Qu. Median Mean 3rd Qu. Max.# 4.170 4.550 5.155 5.032 5.293 6.110可以发现均值大概是5左右,且数据符合正态分布。下面检验和理论值7相比的情况,原假设是等于7,备择假设是不等于7,所以采用双尾检验。
xxxxxxxxxxt.test(weight,mu = 7)# One Sample t-test##data: weight#t = -10.673, df = 9, p-value = 2.075e-06#alternative hypothesis: true mean is not equal to 7#95 percent confidence interval:# 4.614882 5.449118#sample estimates:#mean of x# 5.032通过上述结果看出统计量t为-10.673,自由度是9,p值是2.075e-06<0.05,所以拒绝原假设。
单样本Wilcoxon 符号秩检验
如果样本数据没有通过正态分布检验就要采用单样本wilcoxon符号秩检验进行计算。使用该检验需要满足的条件是样本值均匀地分布在均值两侧。
其R中的函数为wilcox.test()
xxxxxxxxxxwilcox.test(weight,mu=7)通过结果可知拒绝原假设。
独立双样本t-test
两个独立样本是指两个比较的样本之间没有关联,不互相影响。使用双样本独立t-test的前提是两个样本符合正态分布且方差相等。
独立双样本t-test其统计量计算公式为
分别表示两个样本的均值,n表示样本量。表示两个独立样本的方差合并估计,计算公式为
依旧以PlantGrowth数据进行举例,PlantGrowth共有三组数据,在这里取两组进行分析。
xxxxxxxxxx# 查看各组基本信息library(dplyr)tmp1<-group_by(PlantGrowth,group) %>% summarise(count=n(),mean=mean(weight),sd=sd(weight))tmp1# A tibble: 3 x 4# group count mean sd# <fctr> <int> <dbl> <dbl>#1 ctrl 10 5.032 0.5830914#2 trt1 10 4.661 0.7936757#3 trt2 10 5.526 0.4425733进行独立双样本t-test前,首先验证数据是否符合正态分布(Shapiro-Wilk normality test)以及两个样本方差是否相等(F-test)。
关于方差比较
- 两个方差比较通常使用F-test,在R中的函数为var.test(),进行F-test 检验前一定要首先进行正态分布检验,该统计量对于正态分布这个前提条件非常敏感;
- 多个方差进行比较还可以使用Bartlett’s test,该方法同样要求满足正态分布前提条件;
- 如果不能确定数据是否符合正态分布,可以使用Levene’s test 进行检验,其对数据正态分布的要求并非十分严格;
- Fligner-Killeen test 则是一种非参数检验方法,数据可以不满足正态分布;
- 关于方差比较部分内容,在本章不再展开。
xxxxxxxxxxshapiro.test(PlantGrowth$weight[PlantGrowth$group=="ctrl"])shapiro.test(PlantGrowth$weight[PlantGrowth$group=="trt1"])var.test(PlantGrowth$weight[PlantGrowth$group=="ctrl"], PlantGrowth$weight[PlantGrowth$group=="trt1"])# Shapiro-Wilk normality test##data: PlantGrowth$weight[PlantGrowth$group == "ctrl"]#W = 0.95668, p-value = 0.7475### Shapiro-Wilk normality test##data: PlantGrowth$weight[PlantGrowth$group == "trt1"]#W = 0.93041, p-value = 0.4519### F test to compare two variances##data: PlantGrowth$weight[PlantGrowth$group == "ctrl"] and PlantGrowth$weight[PlantGrowth$group == "trt1"]#F = 0.53974, num df = 9, denom df = 9, p-value = 0.3719#alternative hypothesis: true ratio of variances is not equal to 1#95 percent confidence interval:# 0.1340645 2.1730025#sample estimates:#ratio of variances# 0.5397431满足前提条件,继续方差相等的独立双样本t-test
xxxxxxxxxxt.test(PlantGrowth$weight[PlantGrowth$group=="ctrl"], PlantGrowth$weight[PlantGrowth$group=="trt1"], var.equal = T )# Two Sample t-test##data: PlantGrowth$weight[PlantGrowth$group == "ctrl"] and PlantGrowth$weight[PlantGrowth$group == "trt1"]#t = 1.1913, df = 18, p-value = 0.249#alternative hypothesis: true difference in means is not equal to 0#95 percent confidence interval:# -0.2833003 1.0253003#sample estimates:#mean of x mean of y# 5.032 4.661独立双样本Wilcoxon test
当两个样本不满足正态分布时,使用Wilcoxon秩和检验进行非参数检验。
xxxxxxxxxxwilcox.test(PlantGrowth$weight[PlantGrowth$group=="ctrl"], PlantGrowth$weight[PlantGrowth$group=="trt1"], exact = F )# Wilcoxon rank sum test with continuity correction## data: PlantGrowth$weight[PlantGrowth$group == "ctrl"] and PlantGrowth$weight[PlantGrowth$group == "trt1"]# W = 67.5, p-value = 0.1986# alternative hypothesis: true location shift is not equal to 0非独立双样本t-test
在很多试验中需要比较的两组样本往往有关系的,比如一组病人服药前后的变化,每个人以自己为对照。所谓非独立双样本,就是在第一组样本中的每个数据点都和第二组样本中唯一的数据点对应。
非独立样本和独立样本相比,可以显著降低样本量,提高统计的power。其统计量计算公式和单样本t检验一致。
xxxxxxxxxxt.test(PlantGrowth$weight[PlantGrowth$group=="ctrl"], PlantGrowth$weight[PlantGrowth$group=="trt1"], paired = T )# Paired t-test## data: PlantGrowth$weight[PlantGrowth$group == "ctrl"] and PlantGrowth$weight[PlantGrowth$group == "trt1"]# t = 0.99384, df = 9, p-value = 0.3463# alternative hypothesis: true difference in means is not equal to 0# 95 percent confidence interval:# -0.4734609 1.2154609# sample estimates:# mean of the differences# 0.371非独立双样本Wilcoxon test
和单样本类似,如果不符合正态分布则使用非参数检验。
xxxxxxxxxxwilcox.test(PlantGrowth$weight[PlantGrowth$group=="ctrl"], PlantGrowth$weight[PlantGrowth$group=="trt1"], exact = F , paired = T)# Wilcoxon signed rank test with continuity correction## data: PlantGrowth$weight[PlantGrowth$group == "ctrl"] and PlantGrowth$weight[PlantGrowth$group == "trt1"]# V = 37, p-value = 0.359# alternative hypothesis: true location shift is not equal to 0多样本均值分析
在上一节讨论了单样本和双样本均值比较的几种情况,但是很多实验不仅仅有两组样本。进行多组样本之间的均值比较就需要进行单因素方差分析(one-way analysis of variancd)和双因素方差分析(two-way analysis of variancd)
one-way ANOVA
在单因素方差分析模型中样本的组数是任意的但是要求样本之间相互独立,每一组的观测值符合正态分布的同时方差相等,目的是比较每个组的均值。原假设是各组间均值相等(观察的差异有随机误差构成),备择假设是至少有一组和其他组均值不同。数据的变异通常来自于组内部的变异和组间真是变异,其中组内变异一方面由个体差异导致,一方面由实验误差导致,而组间变异则是由不同的处理(因子)导致。如果通过计算,非系统性的变异(组内误差)远大于由不同处理造成的变异(组间变异)如下图b所示,则接受原假设,反之则拒绝原假设,如下图a所示。

其中组间平均平方和=组间MS=BetweenSS/(k-1),k-1为组间自由度,组内平局平方和=组内MS=WithinSS/(n-k),n-k为组内自由度,显著性检验建立在组间平均平方和与组内平均平方和的比值上,且该比值为F分布,自由度为k-1, n-k。
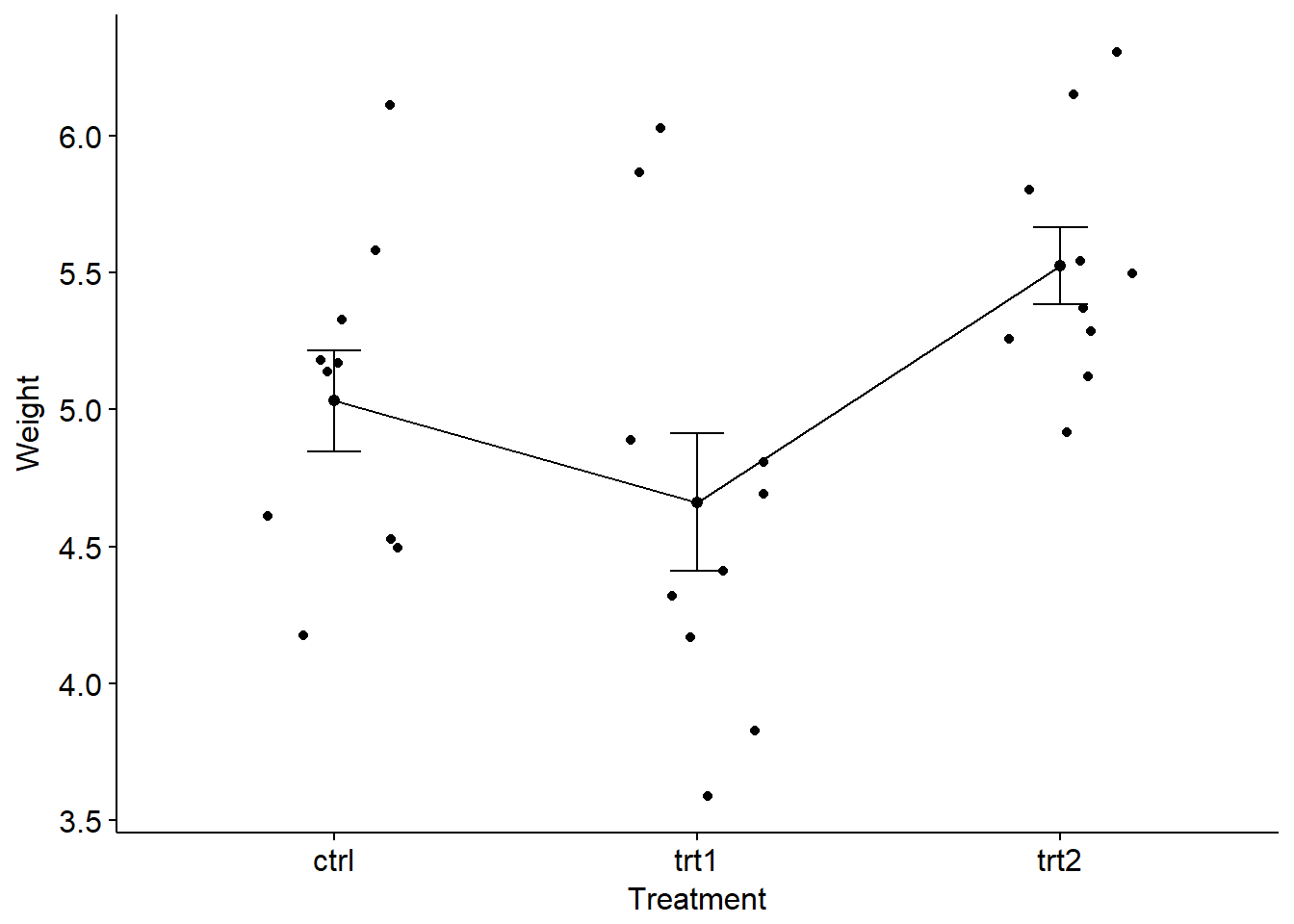
具体到R中,使用R中的PlantGrowth数据集举例。该数据有两个实验组和一个对照组,数据的整体情况上文已经统计过,下面通过box plot进行形象化展示(使用前文提到过的ggpubr包)。
xxxxxxxxxxlibrary(ggpubr)ggboxplot(PlantGrowth, x = "group", y = "weight", color = "group", palette = c("#2072b8", "#ff6a38", "#1e993b"), order = c("ctrl", "trt1", "trt2"), xlab = "Treatment", ylab = "Weight" )ggline(PlantGrowth, x = "group", y = "weight", add = c("mean_se", "jitter"), order = c("ctrl", "trt1", "trt2"), ylab = "Weight", xlab = "Treatment")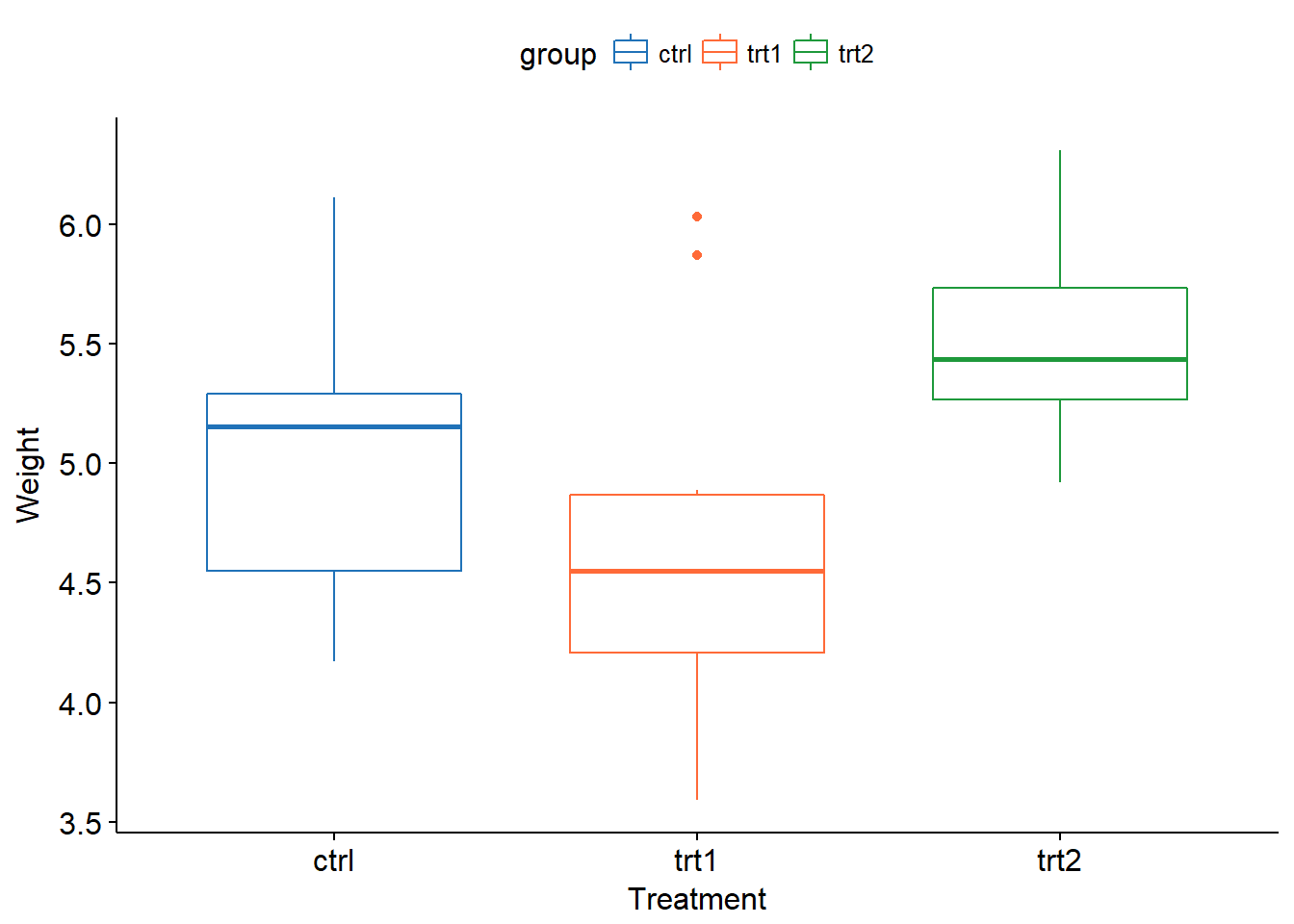
检测正态分布和方差齐性
xxxxxxxxxx#检验方差齐性方法一bartlett.test(weight ~ group, data=PlantGrowth)#检验方差齐性方法二library(car)leveneTest(PlantGrowth$weight~PlantGrowth$group)# Bartlett test of homogeneity of variances## data: weight by group# Bartlett's K-squared = 2.8786, df = 2, p-value = 0.2371## Levene's Test for Homogeneity of Variance (center = median)# Df F value Pr(>F)# group 2 1.1192 0.3412# 27不符合前提条件时
- 如果此步骤检测方差不齐性,可以使用Welch’s ANOVA,
oneway.test(); - 如果不符合正态分布模型,可以使用非参数检验Kruskal-Wallis test,
kruskal.test()和pairwise.wilcox.test()。
进行one-way ANOVA 分析
xxxxxxxxxxpg.aov <- aov(weight ~ group, data = PlantGrowth)summary(pg.aov)# Df Sum Sq Mean Sq F value Pr(>F)# group 2 3.766 1.8832 4.846 0.0159 *# Residuals 27 10.492 0.3886# ---# Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1通过aov(),我们只能判断这三组数据之间均值不相等,但是不知道具体哪组存在差别。为了知道具体的差异情况,可以采用如下三种方法。
当进行任意两组间比较时,需要考虑由family-wise error rate 造成的p值误差,需要对p值进行校正以保证任何两组之间显著性差异的总体概率可以维持在一个固定的显著性水平。常用的校正方法"bonferroni"、"BH"和"fdr"等。
- Pairewise t-test
xxxxxxxxxxpairwise.t.test(PlantGrowth$weight, PlantGrowth$group, p.adjust.method = "fdr")# Pairwise comparisons using t tests with pooled SD## data: PlantGrowth$weight and PlantGrowth$group## ctrl trt1# trt1 0.194 -# trt2 0.132 0.013## P value adjustment method: fdr- Tukey multiple pairwise-comparisons
xxxxxxxxxxTukeyHSD(pg.aov)# Tukey multiple comparisons of means# 95% family-wise confidence level## Fit: aov(formula = weight ~ group, data = PlantGrowth)## $group# diff lwr upr p adj# trt1-ctrl -0.371 -1.0622161 0.3202161 0.3908711# trt2-ctrl 0.494 -0.1972161 1.1852161 0.1979960# trt2-trt1 0.865 0.1737839 1.5562161 0.0120064plot(TukeyHSD(pg.aov))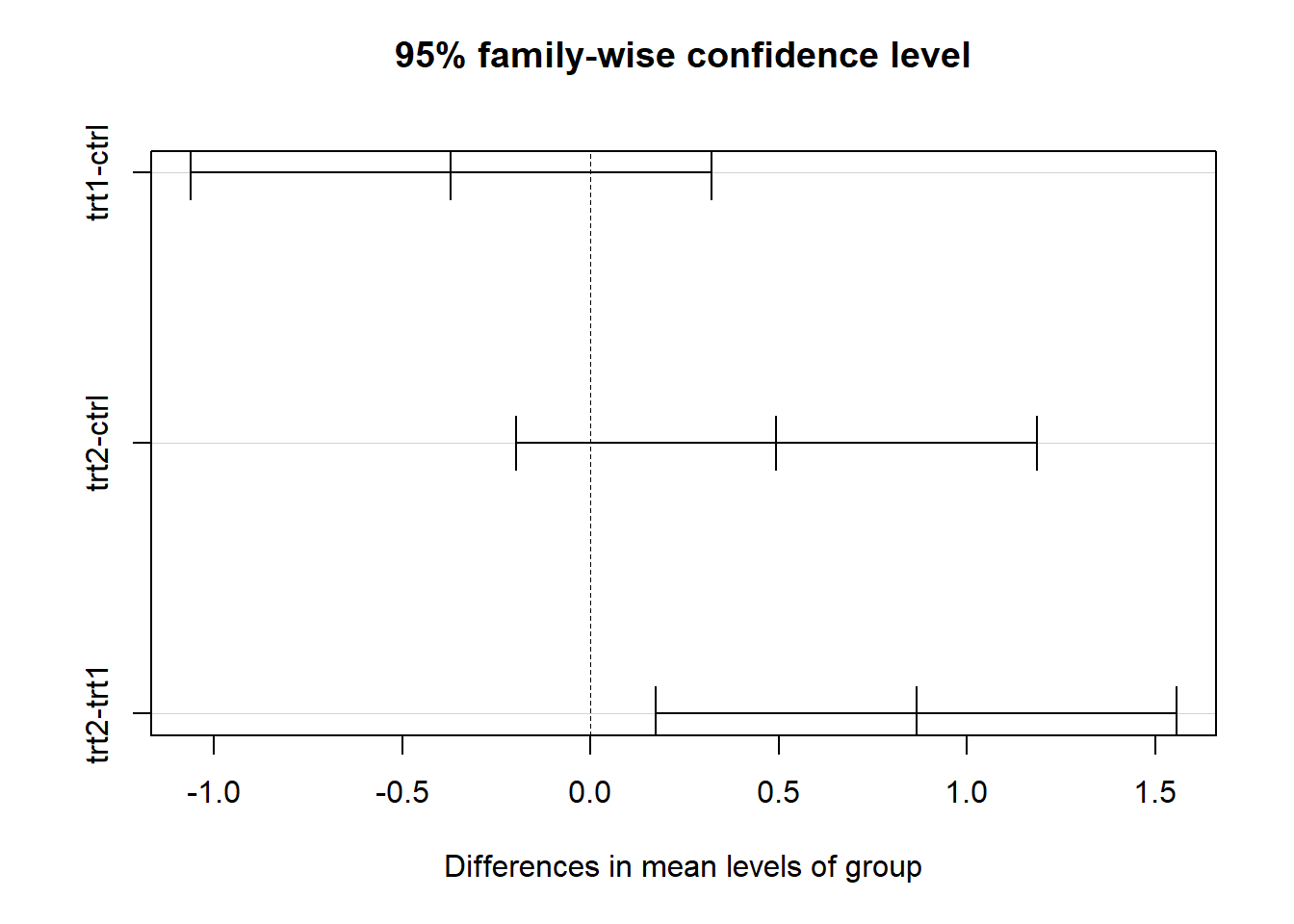
- multcomp包glht()
xxxxxxxxxxlibrary(multcomp)summary(glht(pg.aov, linfct=mcp(group="Dunnett")))summary(glht(pg.aov, linfct=mcp(group="Tukey")))# Simultaneous Tests for General Linear Hypotheses## Multiple Comparisons of Means: Dunnett Contrasts### Fit: aov(formula = weight ~ group, data = PlantGrowth)## Linear Hypotheses:# Estimate Std. Error t value Pr(>|t|)# trt1 - ctrl == 0 -0.3710 0.2788 -1.331 0.323# trt2 - ctrl == 0 0.4940 0.2788 1.772 0.153# (Adjusted p values reported -- single-step method)### Simultaneous Tests for General Linear Hypotheses## Multiple Comparisons of Means: Tukey Contrasts### Fit: aov(formula = weight ~ group, data = PlantGrowth)## Linear Hypotheses:# Estimate Std. Error t value Pr(>|t|)# trt1 - ctrl == 0 -0.3710 0.2788 -1.331 0.391# trt2 - ctrl == 0 0.4940 0.2788 1.772 0.198# trt2 - trt1 == 0 0.8650 0.2788 3.103 0.012 *# ---# Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1# (Adjusted p values reported -- single-step method)#two-way ANOVA
在某些试验中,一个组有两个不同的因子控制,这时需要控制其中一个因子效应后比较另一个因子的效应。此时需要使用双因素ANOVA。如果一个变量对结果的影响依赖于另一个变量的水平,那么这两个变量之间存在交互效应。
下面我们使用R自带数据集ToothGrowth进行说明,该数据集包括药物剂量和喂药方法两个变量。
xxxxxxxxxx# 检测不同条件样本量是否相等table(ToothGrowth$supp,ToothGrowth$dose)# 0.5 1 2# OJ 10 10 10# VC 10 10 10summary(ToothGrowth)# 发现dose并没有当做因子来处理,需要进行转换# len supp dose# Min. : 4.20 OJ:30 Min. :0.500# 1st Qu.:13.07 VC:30 1st Qu.:0.500# Median :19.25 Median :1.000# Mean :18.81 Mean :1.167# 3rd Qu.:25.27 3rd Qu.:2.000# Max. :33.90 Max. :2.000tg <- ToothGrowthtg$dose <- factor(tg$dose, levels = c(0.5,1,2), labels =c("low","mid","high") )# 重新检查summary(tg)# len supp dose# Min. : 4.20 OJ:30 low :20# 1st Qu.:13.07 VC:30 mid :20# Median :19.25 high:20# Mean :18.81# 3rd Qu.:25.27# Max. :33.90进行双因素方差分析
xxxxxxxxxxsummary(tg.aov <- aov(tg$len~tg$supp*tg$dose))## 正确做法# Df Sum Sq Mean Sq F value Pr(>F)# tg$supp 1 205.4 205.4 15.572 0.000231 ***# tg$dose 2 2426.4 1213.2 92.000 < 2e-16 ***# tg$supp:tg$dose 2 108.3 54.2 4.107 0.021860 *# Residuals 54 712.1 13.2# ---# Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1summary(tmp.aov <- aov(ToothGrowth$len~ToothGrowth$supp*ToothGrowth$dose))## 错误做法,不将dose转换为factor,《R语言实战》第一版用到这个例子的时候书上是错误的,第二版已经改正。# Df Sum Sq Mean Sq F value Pr(>F)# ToothGrowth$supp 1 205.4 205.4 12.317 0.000894 ***# ToothGrowth$dose 1 2224.3 2224.3 133.415 < 2e-16 ***# ToothGrowth$supp:ToothGrowth$dose 1 88.9 88.9 5.333 0.024631 *# Residuals 56 933.6 16.7# ---# Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1由上述统计结果可以看出两个变量都对结果有显著的影响,下面将数据可视化展示。
xxxxxxxxxxlibrary(ggplot2)ggplot(tg, aes(x=dose,y=len))+geom_boxplot(aes(fill = dose))+facet_grid(.~supp)ggplot(tg, aes(x=supp,y=len))+geom_boxplot(aes(fill = supp))+facet_grid(.~dose)library(ggpubr)ggboxplot(tg, x = "dose", y = "len", color = "supp", palette = c("#2072b8", "#ff6a38"))ggboxplot(tg, x = "supp", y = "len", color = "dose", palette = c("#2072b8", "#ff6a38", "#1e993b"))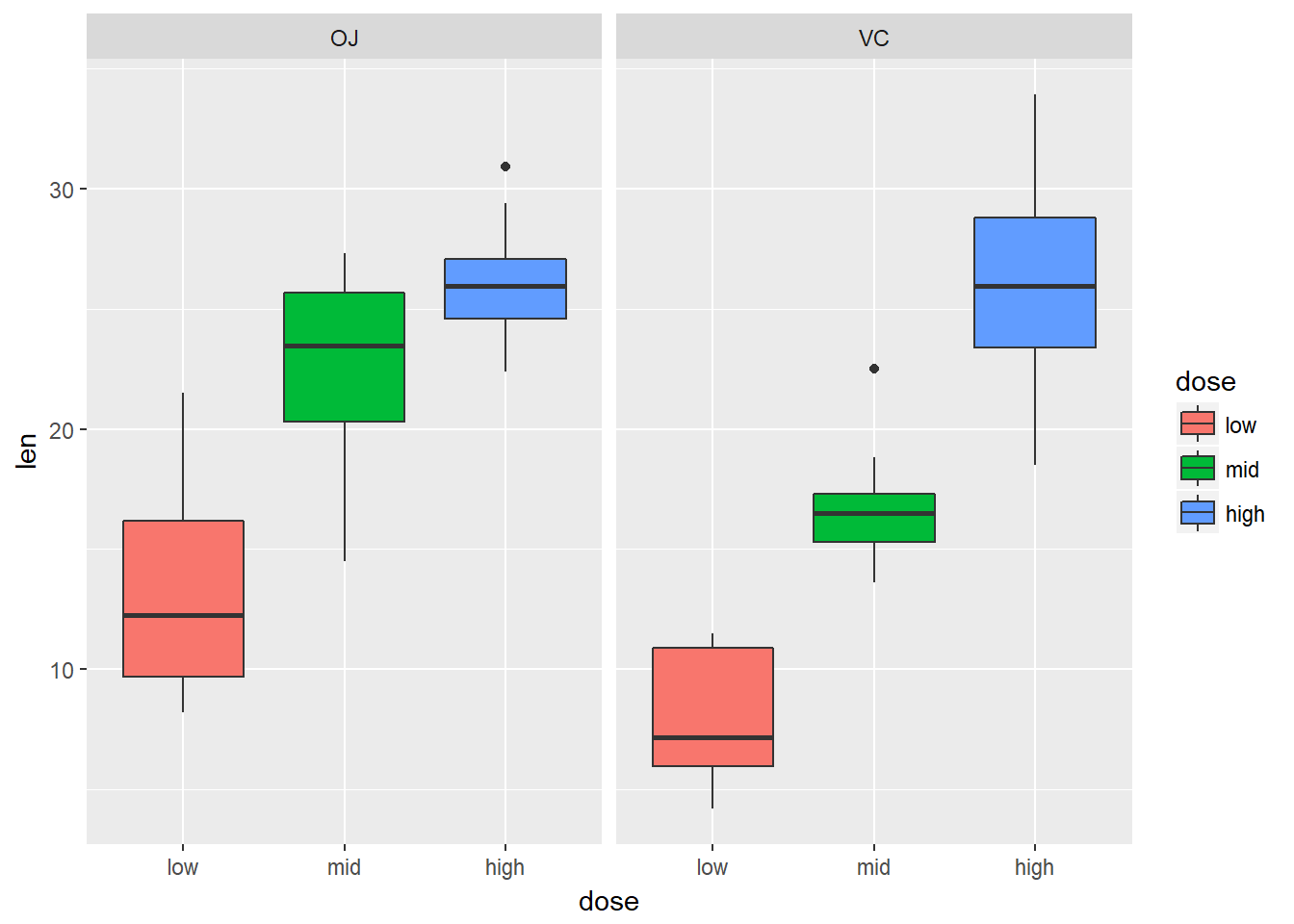
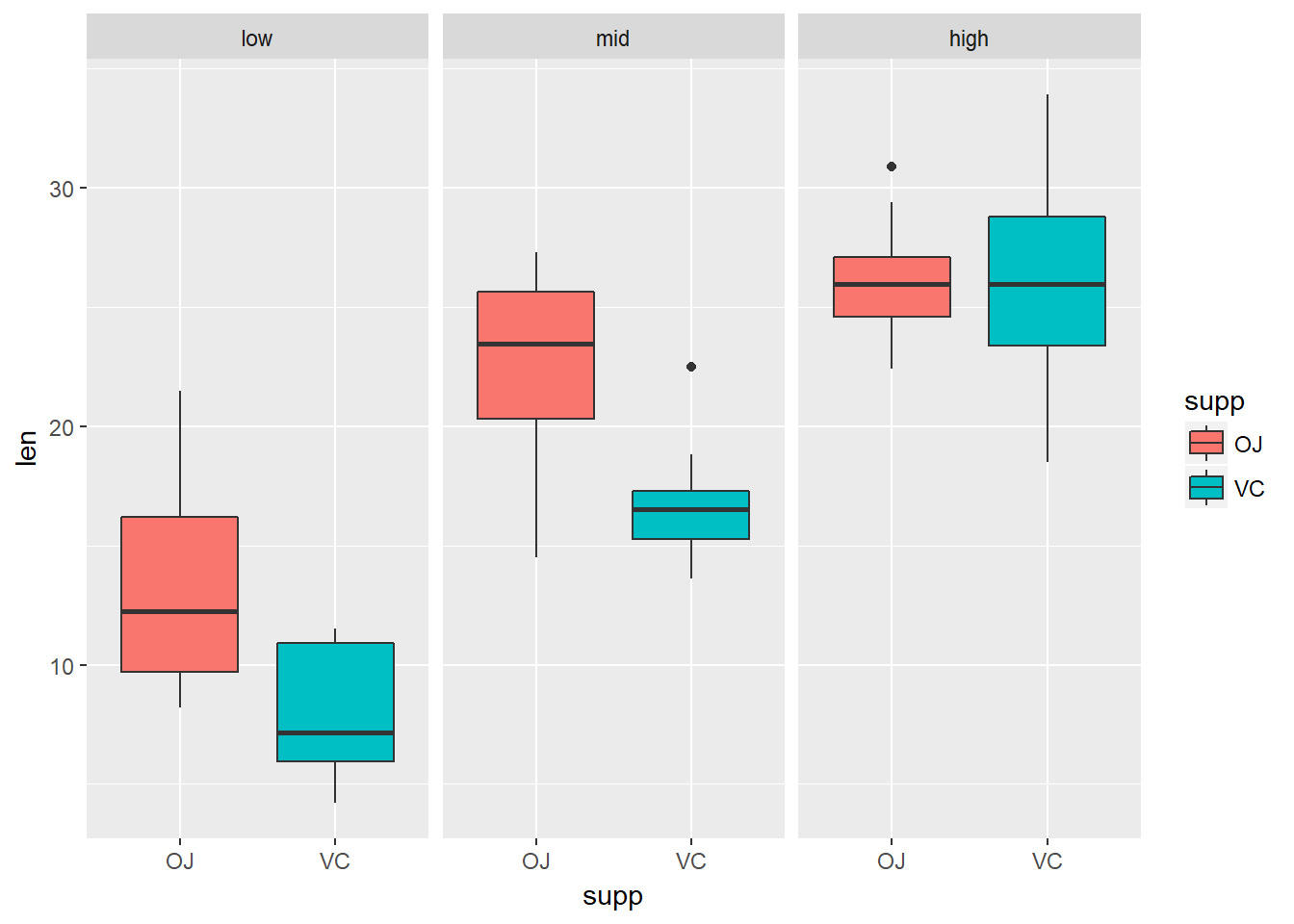
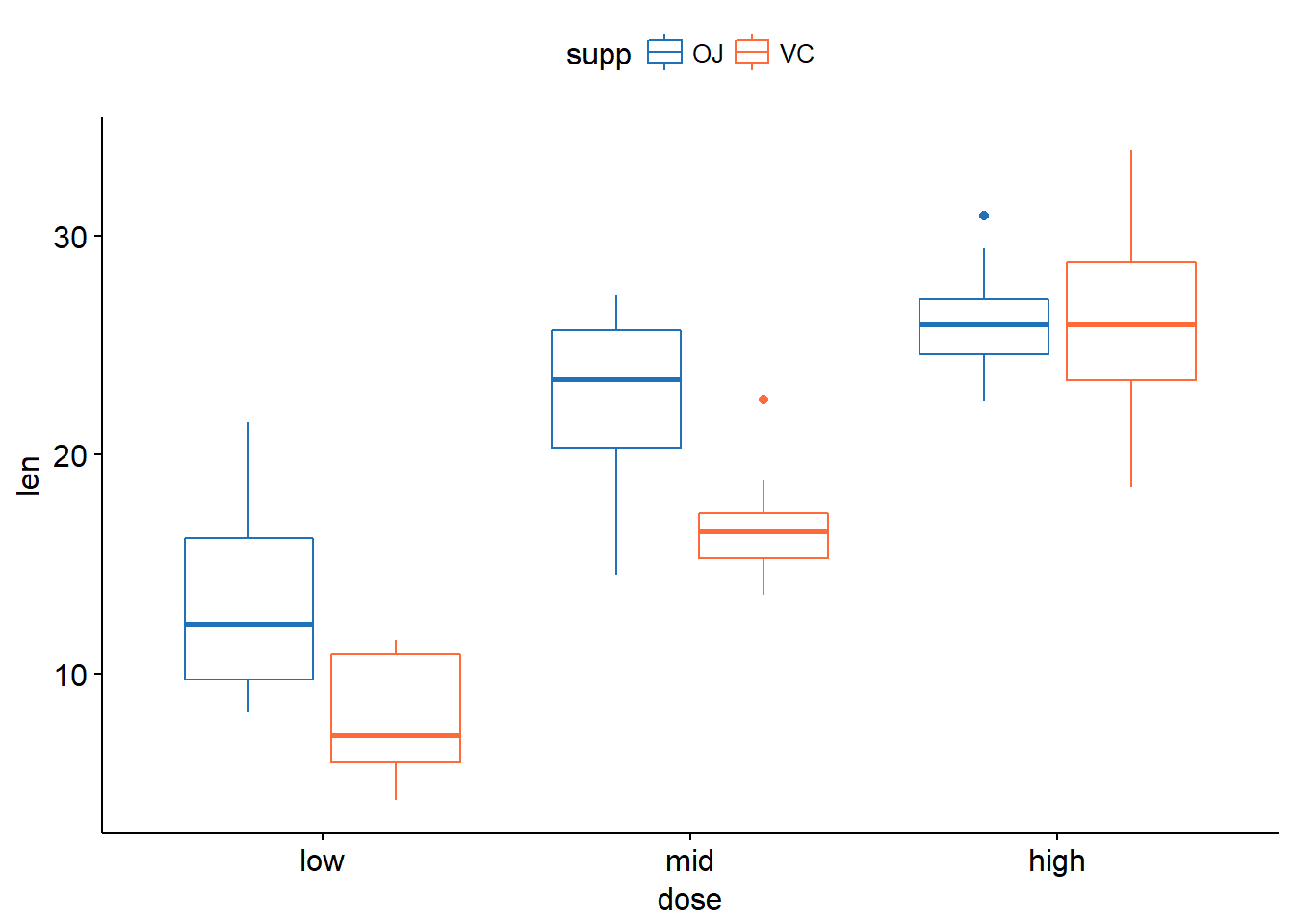
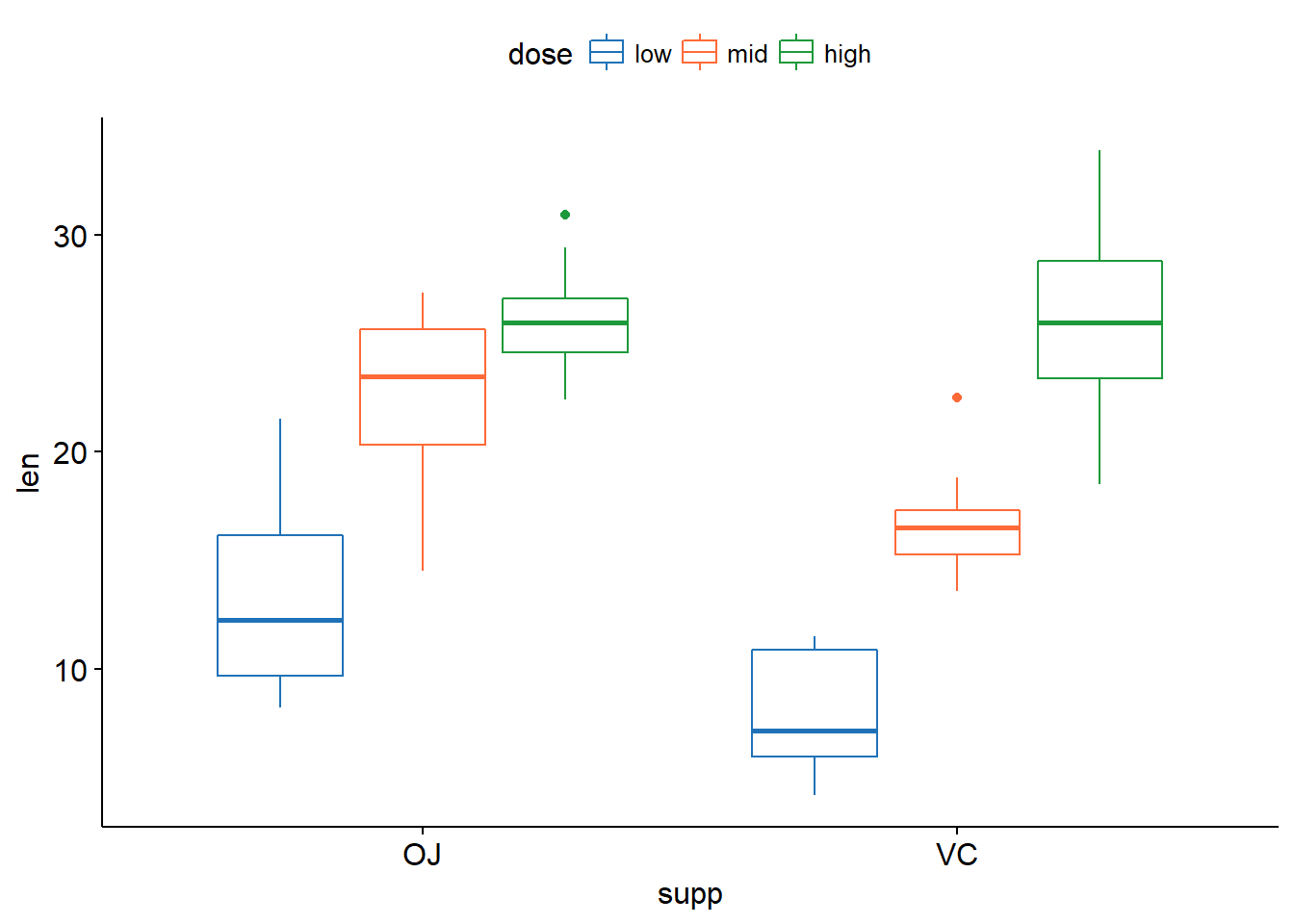
如果想要进行任意两组之间的比较还可以使用下面几种方法:
xxxxxxxxxxTukeyHSD(tg.aov)# Tukey multiple comparisons of means# 95% family-wise confidence level## Fit: aov(formula = tg$len ~ tg$supp * tg$dose)## $`tg$supp`# diff lwr upr p adj# VC-OJ -3.7 -5.579828 -1.820172 0.0002312## $`tg$dose`# diff lwr upr p adj# mid-low 9.130 6.362488 11.897512 0.0e+00# high-low 15.495 12.727488 18.262512 0.0e+00# high-mid 6.365 3.597488 9.132512 2.7e-06## $`tg$supp:tg$dose`# diff lwr upr p adj# VC:low-OJ:low -5.25 -10.048124 -0.4518762 0.0242521# OJ:mid-OJ:low 9.47 4.671876 14.2681238 0.0000046# VC:mid-OJ:low 3.54 -1.258124 8.3381238 0.2640208# OJ:high-OJ:low 12.83 8.031876 17.6281238 0.0000000# VC:high-OJ:low 12.91 8.111876 17.7081238 0.0000000# OJ:mid-VC:low 14.72 9.921876 19.5181238 0.0000000# VC:mid-VC:low 8.79 3.991876 13.5881238 0.0000210# OJ:high-VC:low 18.08 13.281876 22.8781238 0.0000000# VC:high-VC:low 18.16 13.361876 22.9581238 0.0000000# VC:mid-OJ:mid -5.93 -10.728124 -1.1318762 0.0073930# OJ:high-OJ:mid 3.36 -1.438124 8.1581238 0.3187361# VC:high-OJ:mid 3.44 -1.358124 8.2381238 0.2936430# OJ:high-VC:mid 9.29 4.491876 14.0881238 0.0000069# VC:high-VC:mid 9.37 4.571876 14.1681238 0.0000058# VC:high-OJ:high 0.08 -4.718124 4.8781238 1.0000000#上述结果如果感觉过于复杂,可以使用下面下面的形式TukeyHSD(tg.aov,which = "tg$dose")# Tukey multiple comparisons of means# 95% family-wise confidence level## Fit: aov(formula = tg$len ~ tg$supp * tg$dose)## $`tg$dose`# diff lwr upr p adj# mid-low 9.130 6.362488 11.897512 0.0e+00# high-low 15.495 12.727488 18.262512 0.0e+00# high-mid 6.365 3.597488 9.132512 2.7e-06# 或者pairwise.t.test(tg$len,tg$dose)# Pairwise comparisons using t tests with pooled SD## data: tg$len and tg$dose## low mid# mid 1.3e-08 -# high 4.4e-16 1.4e-05## P value adjustment method: holm如果不满足正态分布,则需要使用非参数检验Kruskal-Wallis test,kruskal.test()和pairwise.wilcox.test()。
比例分析
在之前几节内容中提到了均值分析和比较,但有时候我们关心的并不是均值而是比例(proportion)。
单比例检验
对于n比较大(通常为 同时 )的样本来说,根据中心极限定理,样本近似于正态分布,可以使用z检验,其检验统计量计算公式为:
其中,表示观测到的比例,为预期比例,n表示样本量,。
如果样本比较小,则使用二项分布进行统计。
在R中,对于小样本,采用binom.test(),对于大样本使用正态分布近似二项分布,利用prop.test()进行分析。
在单样本比例检验中,我们关心的是具有同种特性的两个群体,在该特性总体中所占有的比例情况。例如,小鼠中公鼠母鼠各有一半,有100只患有某种疾病,其中有公鼠60只,母鼠40只。想知道是否公鼠患病率比母鼠高。在该问题中成功次数为公鼠患病数55,总次数为100,预期比例为50%(公母鼠数量相等)。
xxxxxxxxxxprop.test(60, 100, p = 0.5, alternative = "greater")# 1-sample proportions test with continuity# correction## data: 60 out of 100, null probability 0.5# X-squared = 3.61, df = 1, p-value = 0.02872# alternative hypothesis: true p is greater than 0.5# 95 percent confidence interval:# 0.5127842 1.0000000# sample estimates:# p# 0.6其中,x为成功的次数,n为总测试,p为要测试的概率大小。在结果中,显示了卡方检验的统计量值,自由度和p值和置信区间,最后给出了样本概率估计值。
双比例检验
如果我们已知两组具有不同特性(A和B)样本的样本量和这两组样本中具有某种共同特性(C)的个体数量(也就是知道了C特性各自群体比例和总体比例),想要计算具有C特性的个体在A特性群体和B特性群体中的比例是否一样,就需要用到双比例检验。
当样本数量较小时(所有np和nq都小于5),通常采用非参数检验Fisher Exact probability test 进行分析。当样本力量较大时,我们还是近似使用正态分布z检验来进行预测。
例如,男生500人,女生500人,其中喜欢阅读的男生有400人,喜欢阅读的女生有460人。男生喜欢阅读的比例是否比女生高。我们假设男生喜欢阅读的比例比女生高,则备择假设是男生喜欢阅读的比例比女生低。
xxxxxxxxxx prop.test(x = c(400, 460), n = c(500, 500), alternative = "less")# 2-sample test for equality of proportions with# continuity correction## data: c(400, 460) out of c(500, 500)# X-squared = 28.912, df = 1, p-value = 3.787e-08# alternative hypothesis: less# 95 percent confidence interval:# -1.0000000 -0.0824468# sample estimates:# prop 1 prop 2# 0.80 0.92由结果可知,p<0.05,拒绝原假设,即男生喜欢阅读的比例比女生低。
卡方分布
分布可以通过原假设,得到一个统计量来表示期望结果和实际结果之间的偏离程度,进而根据分布,自由度和假设成立的情况,得出观察频率极值的发生概率(比当前统计结果更加极端的概率)。计算方法是对概率分布中的每一个频率,用期望频数和实际频数差的平方除以期望频数,最后把所有结果相加。得到的统计量结果越大,说明差别越显著,数值越小说明观察和期望的差别越小,当观察频数和期望频数一致是卡方为0。其实就是在比较观测到的比例和期望的比例的关系。
卡方分布就可以用来检验某个分类变量各类的出现概率是否等于指定概率,可以检验数据的拟合优度(指定的一组数据与指定分布的吻合度),也可以用来检验两个变量的独立性(两个变量之间是否存在某种关联)。
在使用卡方检验时,需要的一个参数被称为自由度,指的是独立变量的个数(组数减去限制数)。通常,二项分布已知p,泊松分布已知,正态分布已知和时的自由度是n-1。进行独立性检验时,h行kl列联列表的自由度是。
常用高阶分析方法
回归分析
所谓回归分析(regression analysis),在统计学中有着非常重要的作用,从大的层面来讲指用自变量(解释变量或者预测变量)来预测因变量(相应变量或者结果变量)的方法。比如在几个自变量中找到和因变量更相关的一个,利用自变量和因变量的关系生成等式对因变量进行预测。从小的层面来说,回归的模型非常之多,也非常复杂,例如最小二乘回归,logistic回归和泊松回归等等。
最小二乘回归是最常用到的回归模型,包括简单线性回归(一元一阶),多项式回归(一元多阶)和多元线性回归(多元)。所谓最小二乘法是用一条直线(回归线)拟合一组两变量数据的方法,使得误差平方和(点到直线的距离平方和)最小。对数据进行最小二乘回归分析时,要求数据符合正态分布,独立和同方差性。
简单线性回归可以使用F检验,拟合优度()为回归平方和/总平方和,代表因变量可以被自变量解释的比例。
相关系数(r)描述个数据点与直线的偏离程度,度量回归线和数据的拟合程度。
在R中,lm()是拟合回归模型最简单的函数。针对两个变量,我们一般会先通过简单线性回归进行拟合,通过结果图,根据具体的需要再增加二次项提高预测精度。类似之前提到的ANOVA分析,可以使用summary()函数获取回归模型的详细参数和统计量。
有的时候,我们希望能够让回归模型尽量的简单,既减少自变量的数量还不影响对因变量预测的准确对。这是就可以使用anova()函数,查看是否可以删掉一些回归系数不显著的变量。
如果因变量不符合正态分布或者非连续变量,我们就需要考虑使用广义线性模型来进行拟合。当因变量为类别性变量(二项分布)时可以使用logistic回归,当因变量为计数型(泊松分布)可以使用泊松回归。在R中,这两种方法都可以使用glm()来进行计算。
聚类分析
聚类分析是统计中另一个非常重要的方法,可以帮助我们在多维度的数据中把相似的数据归为子集,每个子集中的数据都具有某种程度的相似性。在具体的生物研究中,聚类可以帮助我们通过表达类似的基因找到功能相关的基因,或者帮助推测未知基因的功能。通常聚类分析也被称作非监督机器学习。
进行聚类分析首先需要计算不同观测值之间的距离,进而生成聚类使用的距离矩阵。 计算距离的方法有很多,选择不同的距离计算方法也会对最终的聚类结果产生很大的影响。常用的距离计算方法有欧氏距离(Euclidean distance)、曼哈顿距离(Manhattan distance)和基于相关性距离(correlation-based distances)的Pearson correlation distances、Spearman's rank correlation、Kendall correlation distance 等等。
另外,在构建距离矩阵之前,往往会对原始的观测数据进行标准化,如计算z score。
总的来说,聚类方法从整体上主要使用的有Partitioning method 和Hierarchical Clustering 两类,其中Partitioning 包括K-means clustering,K-medoids clustering(PAM)等具体方法。
k-means
在k-means算法中,首先我们需要确定数据最终分为几类(k),然后会根据分类数量随机选取k个点最为每个类的初始质心(这也是同一组数据每次聚类的结果都不尽相同的原因),随后其他的点都会通过欧式距离找到分到和自己最近的初始中心。通过一次这样的过程之后,再根据每个集合中的点计算均值得到新的质心,重复之前的过程进行迭代。每次迭代,每个集合中的质心都会重新产生,所有的非质心点再重新分配给新的质心形成集合。如果在一次迭代中,只有非常少的点会发生集合的转移则迭代停止。
需要注意的是,k-means聚类的方法对异常值非常敏感,与之相比PAM要好一些。
在R中,kmeans聚类可以使用stats包中的kmeans()函数,如果想使用K-medoids clustering 可以借助cluster包中的pam()函数。另外,在进行cluster和pca相关的分析中,factoextra 也是一个不错的选择,包括了各种常用的功能。
一个比较完整的k-means聚类分析过程主要包括数据标准化,评估合适cluster数数目,进行计算以及可视化展示等步骤。
Hierarchical Clustering
层次聚类和k-means相比不需要预先设定聚类的数目,最终的聚类结果会以颠倒树状结构显示,不同类别观测值在树的最底层(树叶),越向上节点越少。层次聚类可以细分为Agglomerative Clustering 和 Divisive Hierarchical,前者的思路是从“树叶”向“树干”聚集,后者的思路是从“树干”向“树叶”分裂。一般而言,Agglomerative Clustering 适合在clusters比较少时使用;而Divisive Hierarchical 适合在有大量clusters 时使用,可以选择到那一步停下不再细分。
以Agglomerative Clustering 为例,大致流程是首先将每个对象归为一类,每类仅包含一个对象,计算类与类之间的距离。找到最近的两个类然后合并, 接着计算新类与所有旧类之间的距离。重复之前的过程,直到最后合并成一个类为止。
根据类间距离计算方法的不同,又可以分为五种: single-linkage(类和类两组对象间的最小距离)、 complete-linkage(类和类两组对象间的最大距离)、 average-linkage(两组对象间的平均距离)、centroid linkage 和Ward′s method(在每一步使组内离差平方和增量最小)。
一个比较完整的层次聚类分析过程主要包括数据标准化,计算距离,构建聚类树,确定分组以及可视化展示等步骤。
在R中,计算距离时可以使用dist()函数,构建聚类树可以使用hclust()函数,将树分组可以使用cutree()。当然也可以使用cluster包同时完成上面几个步骤。
以R中数据集USArrests为例,进行聚类分析。
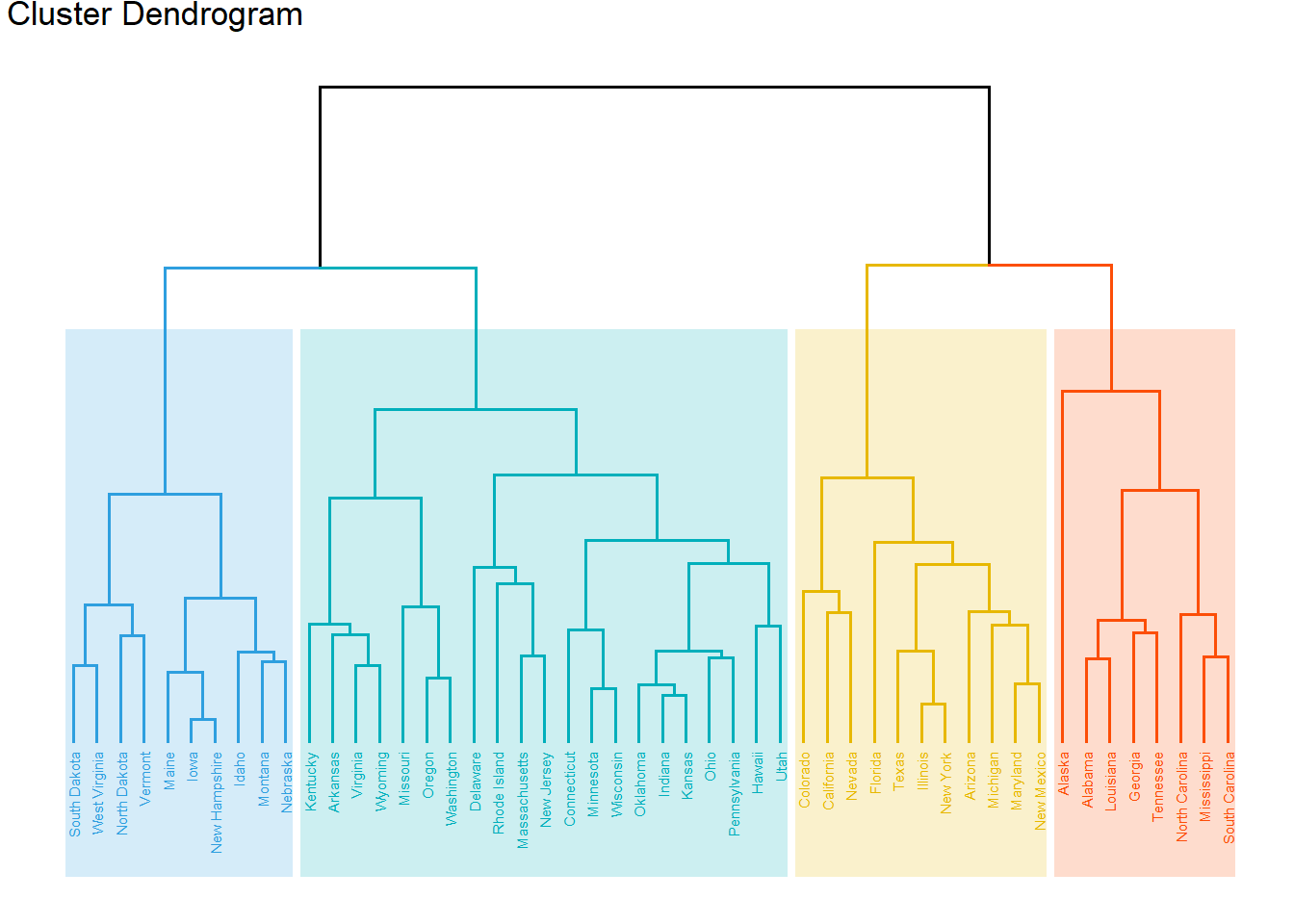
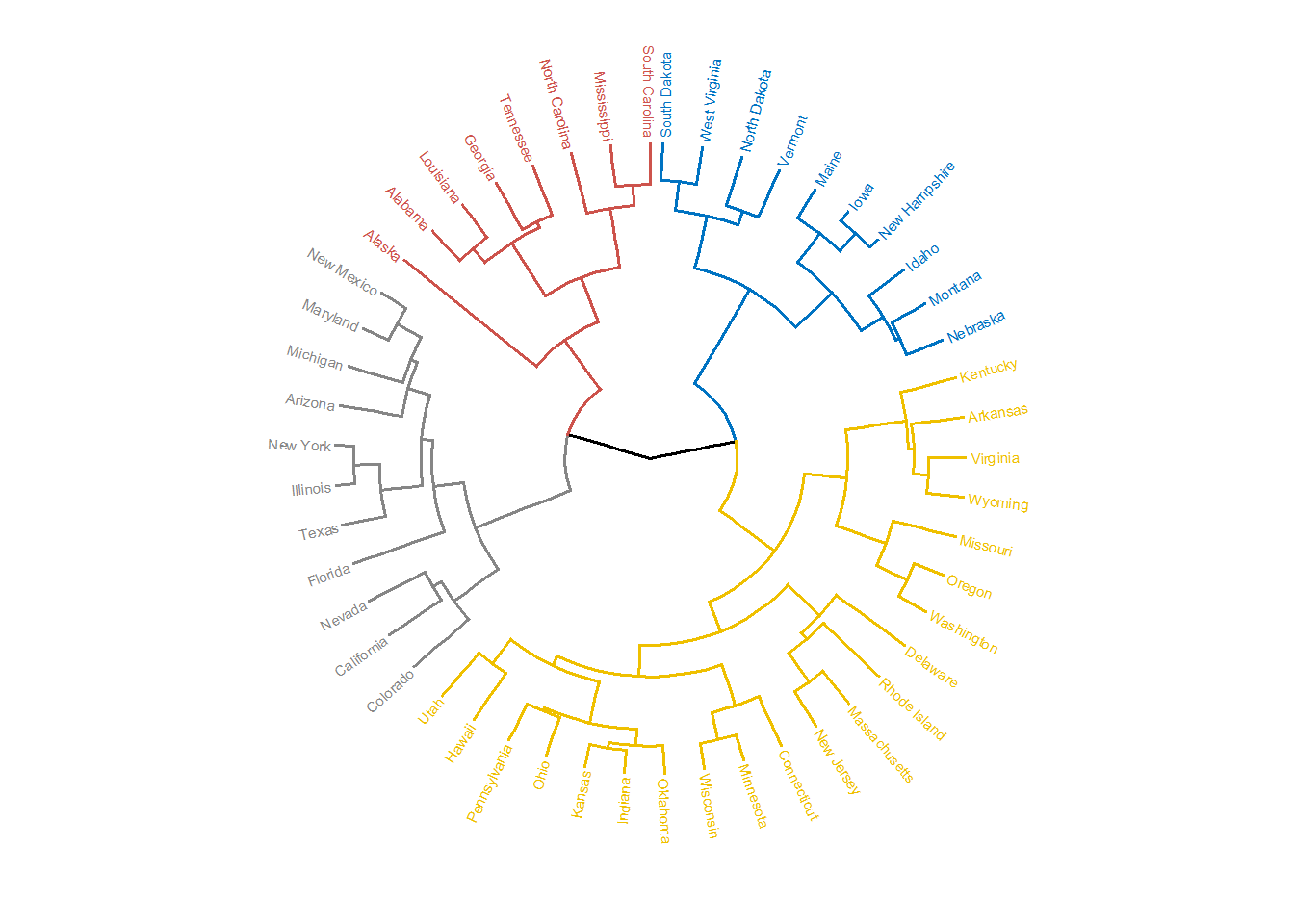
xxxxxxxxxx# 标准化usa_norm <- dist(scale(USArrests), method = "euclidean")# 构建树hc <- hclust(usa_norm, method = "complete")# install.packages(c("factoextra", "dendextend"))# 可视化展示library(factoextra)#基础plot(hc, hang = -1, cex = 0.7)#美化fviz_dend(hc, k = 4, # 分为4类 cex = 0.4, # label大小,防止字不显示完全 k_colors = c("#2E9FDF", "#00AFBB", "#E7B800", "#FC4E07"), color_labels_by_k = TRUE, # color labels by groups rect = TRUE, # Add rectangle around groups rect_fill = TRUE, rect_border = c("#2E9FDF", "#00AFBB", "#E7B800", "#FC4E07"), ggtheme = theme_void() # ggplot2主题 )# 环形fviz_dend(hc, cex = 0.4, k = 4, k_colors = "jco", type = "circular")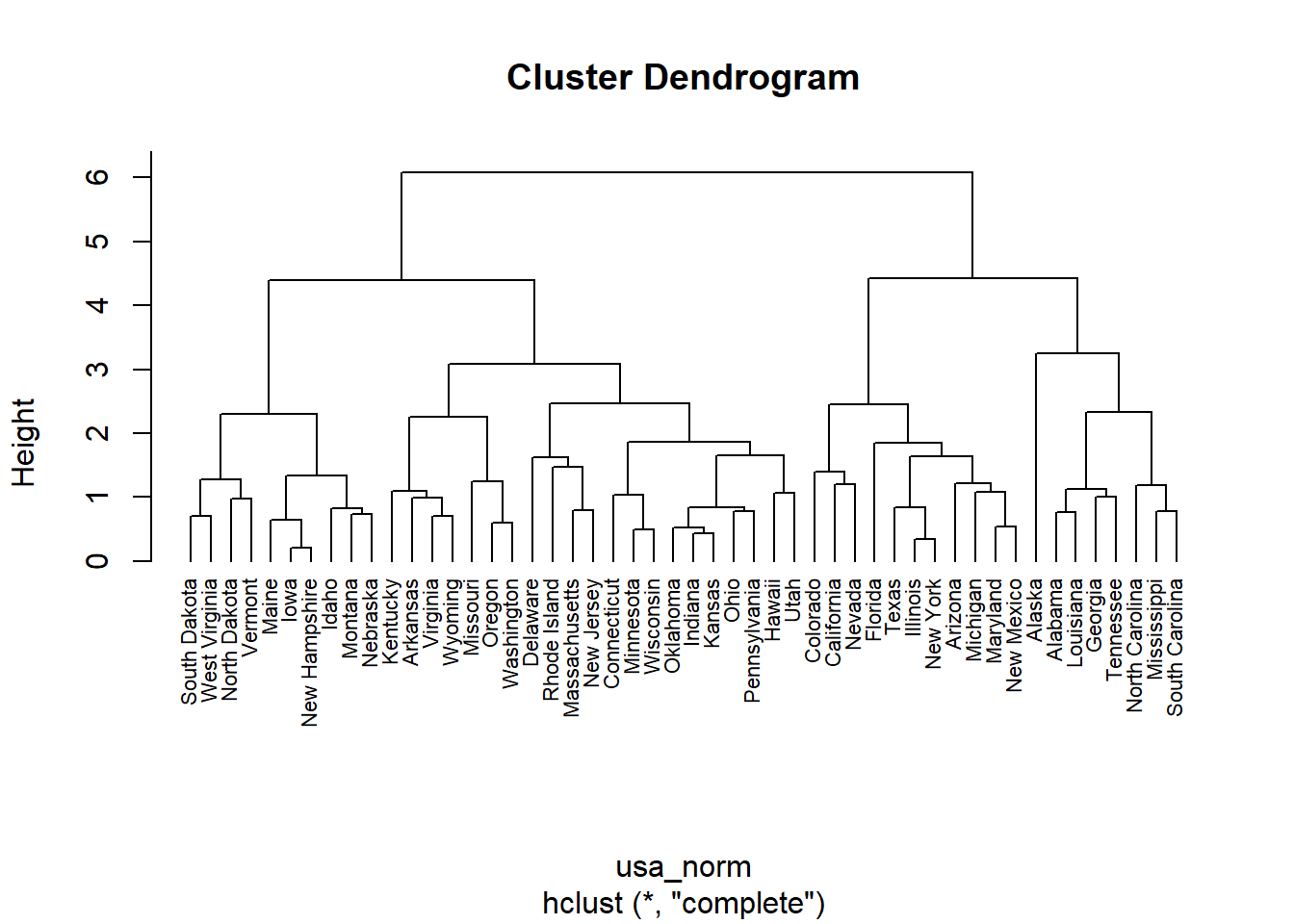
主成分分析
当数据集中变量过多时,会为我们的分析带来很大的不便,在这些变量中很可能存在冗余成分。为了减少冗余变量,降低数据维度,只留下少数能够依旧很好预测因变量的不相关变量,我们可以使用主成分分析(PCA)方法。
在R中,内置函数procomp()可以用来进行PCA分析,以R中数据集iris为例
xxxxxxxxxx# 只支持数值型变量,所以提取原数据集前4列ir_num <- iris[, 1:4]# 进行pca分析ir_pca <- prcomp(ir_num, scale. = TRUE)# 查看主成分信息summary(ir_pca)# Importance of components%s:# PC1 PC2 PC3 PC4# Standard deviation 1.7084 0.9560 0.38309 0.14393# Proportion of Variance 0.7296 0.2285 0.03669 0.00518# Cumulative Proportion 0.7296 0.9581 0.99482 1.00000通过上述结果可以发现,主成分1和2可以解释0.95的变化。